2-лекц. мед.генетикаДокумент Microsoft Office Word. 2 Теориялы аралас сабаты дістемелік зірлемесі Пн Медициналы генетика негіздері Оытушы Маманды Курс Топ
 Скачать 0.71 Mb. Скачать 0.71 Mb.
|

|
№2 Теориялық аралас сабақтың әдістемелік әзірлемесі Пән: Медициналық генетика негіздері Оқытушы: Мамандық: Курс: Топ: Күні: Тақырыбы: Жалпы медициналық патологиядағы тұқым қуалайтын аурулардың орны. Классикалық және классикалық емес типтегі тұқым қуалайтын моногенді аурулар. Сабақтың мақсаты: Білімділік: Оқушы меңгеруі және білуі тиіс: хромасомалардың әртүрлі типтерін дифференциялау және адамның қалыпты және патологиялық кариотипін ажырата білу Тәрбиелік: Құрал-жабдықтармен оқулықтарды таза ұстауға үйрету; Өз-ісіне жауапты болуға, ұқыптылыққа, шапшаңдыққа баулу; Оқу орнын қадірлеуге тәрбиелеу. Дамытушылық: Тәжірибе сабағымен байланыстыра білуге үйрету; Өз пікірін дұрыс жеткізуіне көңіл бөлу; Тақырыпты дұрыс баяндай білуіне назар аудару; Сабақтан алған ақпараттарды талдап үйрену; Алған білімдерін басқа пәндерде, тәжірибе сабағында қолдана білу; Ғылыми-оқу және анықтамалық әдебиеттермен өз бетінше жұмыс істеп білуге үйрету; Сабақтың уақыты: 90 минут (2 сағаттық) Сабақты өткізу орны: Сабақтың түрі: Аралас сабақ Сабақты жүргізу әдісі: Көргізбелі, ауызша, жазбаша, тірек-сызба. Пән ішілік байланыс: Полигенді және мультифакторлы аурулар Пән аралық байланыс: Жалпы биология, Медициналық биология және генетика, Молекулярлы биология Сабақтың жабдықталуы: Материалдық: молекулалық биология және генетика оқулығы, күн тақырыптық жоспар, оқу жұмыс бағдарламасы, тақырыптық жоспар, тесттер, әдістемелік әзірлеме Техникалық-программалық: СД диск, компьютер, интерактивті тақта Пайдаланылатын әдебиет: Негізі: Б.Бегімқұл, С.Төлегенов Медициналық генетика негіздері Астана 2008 Қосымша: Е.Ө. Қуандықов, С.А.Әбилаев Медициналық Биология және Генетика Алматы. Оқушы білуі тиіс: хромасомалардың әртүрлі типтерін дифференциялау және адамның қалыпты және патологиялық кариотипін ажырата білу Сабақтың хронокартасы. 1.Ұйымдастыру кезеңі -5 минут. 2.Оқушылардың білімін тексеру -25 минут. 3. Жаңа тақырыпты түсіндіру -40 минут. 4.Жаңа тақырыпты пысықтау -10 минут. 5.Сабақты қорытындылау -5 минут. 6. Үйге тапсырма -5 минут. Сабақтың барысы: І. Ұйымдастыру кезеңі - 5 минут. Оқытушы оқу бөлмесінің тазалығына, оқушылардың киіміне назар аударады. Оқушылардың сабаққа қатысуы мен дайындығын тексеріп, оларды сабақтың бағдарламасы және мақсатымен таныстыру. ІІ. Оқушылардың білімін тексеру - 25 минут. Дәріс сабағында оқушылардан өтілген тақырыпты сұрайды. Оқушылардың білім деңгейін фронталь сұрақтар қою немесе тестілеу арқылы тексеріледі. Фронталь сұрақтар: Адам патологиясындағы генетикалық факторлардың рөлін зерттейтін жалпы генетика ғылымының маңызды бөлімі қалай аталады ? Фермент дегеніміз не ? Предиктивтік медицина дегеніміз ? Коллагендік аурулар ненің өзгеруіне байланысты пайда болады? Гемоглобинопатиялар қандай ауруларға жатады. Даун синдромын анықтаған ғалымдар ? ІІІ. Жаңа тақырыпты түсіндіру -40 минут Жаңа сабақтың жоспары: Жұлын амиотрофиясы – жиілігі Зат алмасудың тұқым қуалайтын аурулары Жұлын амиотрофиясы - жиілігі жағынан аутосомды-рецессивті аурулардың iшіндe муковисцидоздан кейн, eкiнші орын алатын ауру. Бұл ауру нейроналдық апоптоз ингибиторы — геминнің бұзылуы салдарынан дамиды және бұлшықет әлсіздігі, бұлшықет атрофиясы т.б. нейромоторлық бұзылыстармен сипатталады. Бұл аурудың 3 формасы белгілі: I тип- Вердинг-Гофман ауруы; II тип - созылмалы жұлын (спинальная) амиотрофиясы; III тип-Кугельберг-Веландер ауруы. I тип - Вердинг-Гофман ауруы - нәрестелерде алғашқы 6 айлығында дамиды және өте қатал, зілді болуымен сипатталады. Ауру белгілері құрсақ ішінде даму кезінде — құрсақ ішіндегі баланың өте баяу қимылдауы (қозғалуы) арқылы байқалады. Туылғаннан кейін бұлшықет гипотониясы, сіңір рефлекстерінің ceмyi т.б. байқалады. Нәрестелер басын ұстай алмайды және аударыла алмайды. Алғаш аяктарының төменгі бұлшықеттері зақымданып, әpi қарай дененің жоғары бөлімдеріне таралады. Тынысалу бұлшықеттерінің (қабырға аралық және көкет) зақымдануы көкірек клеткасының деформациялануына алып келіп, сколиоз және омыртқа жотасының көкірек бел бөлімінің кифозына ұласады. Аурудың дамуы барысында зақымдану кеңірдек және жұткыншаққа таралады. Аурудың туа біткен формаларында нәрестелер жүрек және тыныс алу мүшелерінің қызметінің жетімсіздігі және инфекция салдарынан бip жасқа дейін дүние салады. 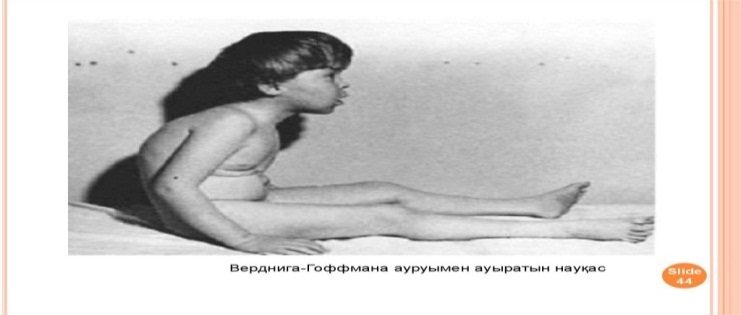  II тип - жұлын (спинальная) амиотрофия ауруының созылмалы формасы — 6-18 айдан кейін дамиды және аса бip ауыр болмайды. Нәрестелер алғашқы кездері қалыпты дамиды — басын ұстай алады, отырады, бipaқ өз бетінше жүре алмайды. Бұл формаға — қол-басы, тіл, иық және жамбас белдеулері бұлшықеттерінің дірілдеп тартылуы, омыртқа жотасының деформациялануы т.б. белгілер тән. Сырқаттардың тipшiлiк ұзактыгы 10-12 жыл. III тип — Кугельберг-Веландер ауруы — 18 айдан 20 жас аралығында дамиды. Жамбас белдеуінің шеткі бұлшықеттері ең алғаш зақымданады. 2-7 жастағы балалар жүрген, жүгірген, баспалдақпен көтерілген кездерде қиналады. Қолдың және иық белдеуінің шеткі бұлшықеттерінің зақымдануы ауру белгілері байқалғаннан кейін бірнеше жылдан соң зақымданады.   Адреногенитальдық синдром - бұл аурудың орташа жиілігі жаңа туылған нәрестелердің 1:12000 тең. Бұл ауру патогенезінің нeгiзi болып бүйрек үсті бездің жыныс және стероидтық гормондардың синтезделу тізбегінің (каскад) бip ферментінің белсенділігінің бұзылуы саналады. Адреногенитальды синдромның ең кең таралған формасы (90-95%) -прогестеронның дезоксикортикостеронға жэне 17-гидроксипрогестронның 11-дезоксикортизолға айналуын катализдейтін 21-гидроксилаза (цитохром 450 с21) ферментінің жетімсіздігіне (дефицит) байланысты дамиды. Оның екі классикалық клиникалық формасы белгілі-сольтерленуші және жай верильдік формасы. Сольтерленуші формасы — ферменттің толық жетімсіздігі (дефицит) жэне тұз айналымының бұзылуымен сипатталады. Патологиялық үдеріс ренин-альдостерон жүйесінде байқалады. Аурудың клиникалық көpiнicтepi нәрестенің туылған күнінен бастап байқалады. Ол - кұсу, шеткі қанайналымның жетіспеушілігі, ұйқышыл болуы, азу т.б. күйлерінде дамиды. Ағзаның сусыздануы нәрестенің шөлдеуіне алып келеді және ол ана емшегін жиі сорады. Биохимиялық зерттеулер — гиперкалиемия, гипонатриемия, ацидоз болатынын көрсетеді. Жай верильдеуші формасы - вирилденудің үделеуі, соматикалық. дамудың жеделденуі, бүйрекүсті бездің гормондарды бөліп шығаруының жоғарылауымен сипатталады. Жаңадан туылған қыз балалардың сыртқы жыныс мүшелері, ерте даму нәтижесінде, ересек әйелдердің сыртқы жыныс мүшесінe ұксас болады, яғни верилденеді. Мұныц негізгі ceбeбi - гипофизде аденокортикоидтық гормондар концентрациясының көбеюі салдарынан адреногормондардың өте көп мөлшерде синтезделуі болып табылады. Коллагенопатиялар – дәнекер ұлпалардың маңызды құрылымдық бөлімдері – коллагендердің синтезделуін және ыдырауын бұзатын мутацияларға байланысты дамитын тұқым қуалайтын аурулардың үлкен бір тобы. Коллагендер – адам ағзасының ақуыздар массасының үштен бір бөлігін құрайды. Коллагендердің 40% теріде, 50%- қанқа құрылымдарында, 10% ішкі мүшелер стромаларында кездеседі. Коллагендік ақуыздар 1000 аминқышқылдарынан құрылған ширатылған 3 полипептидтік 𝛼 – тізбектерден тұрады.Аминқышқылдар құрылымымен ерекшелінетін бірнеше 𝛼 – тізбектер белгілі. Коллагендердің әртүрлі типтері біркелкі не әртүрлі 𝛼 – тізбектерден құралған гомо-не-гетеромерлі болуы мүмкін. Қазіргі кезде коллагендердің 19 типі анықталған. Олардың гендері 14 хромосомада орналасқан, 30-дан астам ақуыздарды қалыптастырады. 1,2,3,5,9-шы типті коллагендер дәнекер құрылымдарда кең таралған фибриллалық коллагендерде, 4 – ші типті коллаген базальдық мембранада кездеседі, 10- шы типті коллагенкөз бұршағының және көздің торлы қабатының құрылымдарын, 11, 12, 14 – ші коллагендер әртүрлі ұлпалардың үлкен коллагендік фибриллаларының құрылымдарын қалыптастырады. 6 типті коллаген жұмсақ ұлпалар мен сіңірлердің микрофибриллдерін пайда етеді, 7 – ші типті тері дермасымен эпидермис құрылымдарының өзара байланысуында бекіндіруші рөл атқарады, 13,17 – ші типті коллагендер – трансмембраналық болып табылады, ал 15,18 – эндотелий құрамына кіреді. Коллагендік аурулар – транскрипция, трансляция, процессинг, тасымалдау үдерістерін бақылайтын гендер және әртүрлі типті коллагендердің қалыптасуына қатынасатын гендер мутациясы негізінде дамуы мүмкін. Элерс-Данло синдромы - түрліше жолдармен тұқым қуалайтын дәнекер ұлпаның гетерогенік аурулар тобы. Оның кeйбіp формаларын 1657 жылы Голландия оташысы Д.ван Мекрен сипаттап жазған. 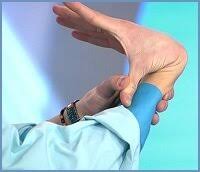 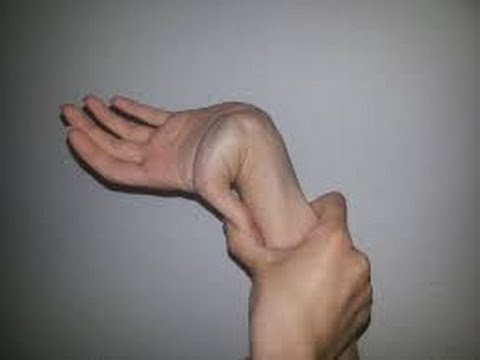 Кейінірек Э.Элерс (1901) және Х.АДанло (1908) бұл синдромды толығырақ зерттеген. Элерс-Данло синдромының ең негізгі белгілерінің бірі-коллаген гендерінің мутациялануы салдарынан коллаген синтезінің бұзылуы негізінде дәнекер ұлпаның туа біткен өте күшті (шектен тыс) созылмалы болуы саналады . Қaзipгi кезде Элерс-Данло синдромының 10 типі анықталған. Олардың 1-4,7,8 типтері аутосомды-доминантты, 6-типi аутосомды-рецессивті, 5,9 типтері Х-тіркескен жолдар арқылы тұқым қуалайды. Элерс-Данло синдромының симптомдар кешені оның әртүрлі типтерінде түрліше қайталана береді, сондықтан олардың бәрімен де танысқан жөн. Олар: 1 Tepici: өте күшті созылғыш, мақпалдай жұмсақ болуы, сынғыш, жиі қанағулар, қоңыр-қара сепкілдердің (20-дан астам), көптеген тыртықтардың болуы; Буындары: шына шақтың-(5 саусқтың) 90° Kepi кайырылуы, шынтақ буынының, тізе буынының 10°-қа еркін кepi қайырылуы, буындардың жеп-жеңіл тайып шығуы, жалпақ табан; 3. Көздері-птоз, тор қабатыныңтүсіпқалуы, көзалмасыныңжыртылуы; 4. Құлақтары: өтекүштісозылуы; 5.Ticтepi: iшiнapa адентия, артықтістердіңболуы, парадантоз, кариес; 6.Аяк-колдары: варрикоздыквеналар, жіліншікте тepi асты түйіндердің болуы, жалпақ табан; 8 Жүрек: аритмия, вегетоқантамырлық (вегетососудистая) дистония, жүрекше-қарынша аралық қалпақша пролапсы; 9. Iшкi мүшелер: асқазан, бүйрек және жатыр птозы; 10. Ми: ми сосудтарының аневризмы, қанқұйылулар.  Зат алмасудың тұқым қуалайтын аурулары адамдардың ең жиі кездесетін және жақсы зерттелген моногендік аурулар тобы болып табылады. Көптеген аурулардың клиникалық көpiнici субстраттарды (заттарды)ыдыратуға және тасымалдауға қатынасатын не жасуша рецепторлары қызметтерін атқаратын ферменттердің кaтaлиздeyшi қызметтерінің жойылуына негізделеді.  Зат алмасудың тұқым қуалайтын ауруларының жіктелуі әлі күнге дейін толық қалыптаспаған, дегенмен оларды төмендегідей топтарға топтастырады. Аминқышқылдарының алмасуының тұқым қуалайтын аурулары аминоацидопатиялар (альбинизм, фенилкетонурия, терозинимия т.б.) . Көмірсулардың алмасуының тұқым қулайтын аурулары глюкозуриялар (галактуземия, глюкогеноздар) . Липидтер алмасуының тұқым қуалайтын аурулары - липидоздар (жанұялық гиперхолистеролемия, сфинголипидоздар, лейкодистрофиялар) Стероидтық гормондардың алмасуының тұқым қуалайтын аурулары(ареногенитальдық синдром). Эритрондардың алмасуның тұқым қуалайтын аурулары (гемолитикалық анемиялар- қаназдылық) Аминқышқылдарының алмасуының бұзылуы нәтижесінде дамитын көптеген тұқым қуалайтын аурулар белгілі.Ең жиі кездесетін және жақсы зерттелген аминоацидопатиялар – фени лаланин және тирозин аминқышқылдарының алмасуның бұзылуына байланысты. Фенилаланин - ағзада синтезделмейтін , тек ас құрамында ағзаға жеткізілетін , алмастыруға болмайтын аминқышқылы.Бауыр жасушаларында экспрессияланатын фенилгидроксилаза көмегімен ол тирозинге айналады.Тирозиннің әрі қарай алмасулары бірнеше жолдармен жүзеге асады. Фенилкетонурия – фенилгидроксилаза ферментінің белсенділігінің жеткіліксіз болуы нәтижесінде дамиды. Оны 1934 жылы көрсеткен. Кейін Пенроуз бұл аурудың аутосомды – ресессивті тұқым қуалайтындығын анықтаған. ФКУ орташа жиілігі – жаңа туылғандардың 1:10000 тең.   Альбинизм – сырқаттар терісінде, шаштарында және көз құрылымдарында меланиннің болмауы не жеткіліксіз мөлшерде болуы нәтижесінде дамитын бір топ тұқым қуалайтын аурулар. Меланин меланоциттер деп аталатын жасушалар субпопуляцияларында өндіріледі. Шаш баданаларында тирозиназаның болуы – болмауына байланысты екі түрлі альбинизм ауруы дамиды.  І типті – тері альбинизмі аутосомды – ресессивті тұқым қуалайтын ауру, жиілігі жаңа туылғандардың 1: 20000 тең. Мутациялардың көпшілігі – жеке нуклеотидтің алмасуы. Фермент белсенділігіне қарай аурудың екі нұсқасы белгілі: 1 нұсқасында фермент белсенділігі О-ге жақын, яғни белсенділіктің болмауымен сипатталады. Нәрестенің терісі, шаштары сүттей аппақ, қалдары болмайды, сырқаттар еш уақытта күнге күймейді. Екінші нұсқада – тирозиназа ферментінің белсенділігі 20-30% деңгейде болады; клиникалық симптомдары – терінің, шашатардың пигменттелуі (боялуы) - әлсіз болады. ІІ типті көз-тері альбинизмі– бұл афро-американдықтарға тән, жиілігі-1:4000 нан 1:1100 ге дейін болатын ауру. Бұл ауру-да аутосомды – рецессивті жолмен тұқым қуалайды. Оның даму себебі – 15 хромосомада орналасқан және 25 экзоннан тұратын мембраналық интегралдық ақуыз –Р генінің мутациялары болып табылады. Бұл ауру нұсқасына - тері, шаштардың пигменттелуінің төмендеуі тән. Сырқаттар терісінің түсі- ақшыл дан қоңырға дейін өзгереді.
ІV. Жаңа тақырыпты пысықтау – 10 минут. Бақылау сұрақтар: Жиілігі жағынан аутосомды-рецессивті аурулардың iшіндe муковисцидоздан кейн, eкiнші орын алатын ауру қалай аталады ? Сипаттама бер. I тип- Вердинг-Гофман ауруы; II тип - созылмалы жұлын (спинальная) амиотрофиясы; III тип-Кугельберг-Веландер ауруы. Түрліше жолдармен тұқым қуалайтын дәнекер ұлпаның гетерогенік аурулар тобы синдромы қалай аталады? Қысқаша негізгі түсінік жаз. | |||||||||||||||