Оразалиева Айжан АНЕСТЕЗИН. Анестезин
 Скачать 58.69 Kb. Скачать 58.69 Kb.
|

|
Анестезин 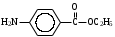 Эмпирическая формула: С9Н11NO2 Молекулярная масса: 165,1 Синонимы – этилбензоат, анасталгин, бензокаин и др. Свойства: белый, кристаллический порошок без запаха, слабо горького вкуса, вызывает скоропроходящее чувство онемения на языке. Растворим в спирте (1:5), жирах, жирных маслах, эфире, хлороформе, дихлорэтане, хуже в толуоле, бензоле. Трудно растворим в разведенной соляной кислоте, лучше – в соляной кислоте концентрацией выше 4,5–5 %. Очень мало растворим в воде, чуть лучше – в кипящей воде. Тпл = 89–91,5 °С. Содержание в препарате не менее 99,9 %. Применяется как высокоэффективное местноанестезирующее средство. Описание. Белый кристаллический порошок без запаха, слабо горького вкуса. Вызывает на языке чувство онемения. Растворимость. Очень мало растворим в воде, легко растворим в спирте, эфире, хлороформе, трудно растворим в жирных маслах и разведенной соляной кислоте. Подлинность. Препарат дает характерную реакцию на ароматические первичные амины (стр. 743). 0,05 г препарата нагревают с 5 мл раствора едкого натра и приливают 0,1 н. раствор йода до неисчезающего желтого окрашивания; появляется запах йодоформа. 0,05 г препарата растворяют в 2 мл воды с 5 каплями разведенной соляной кислоты и прибавляют 2 мл раствора хлорамина. Через 2-3 ми-нуты добавляют 2 мл эфира и взбалтывают; эфирный слой окрашивается в оранжевый цвет. Температура плавления 89-91,5°. Прозрачность и цветность раствора. Раствор 1 г препарата в 10 мл нейтрализованного по фенолфталеину спирта должен быть прозрачным и бесцветным. Кислотность. К полученному раствору прибавляют 3 капли раствора фенолфталеина. Розовое окрашивание должно появиться от прибавления не более 0,1 мл 0,05 н. раствора едкого натра. Хлориды. Раствор 1 г препарата в 10 мл спирта должен выдерживать испытание на хлориды (не более 0,002% в препарате). Органические примеси. 0,5 г препарата растворяют в 5 мл концентрированной серной кислоты. Окраска полученного раствора не должна быть интенсивнее эталона № 5а. Сульфатная зола и тяжелые металлы. Сульфатная зола из 0,5 г препарата не должна превышать 0,1% и должна выдерживать испытание на тяжелые металлы (не более 0,001 % в препарате). Количественное определение. Около 0,2 г препарата (точная навеска) растворяют в 10 мл воды и 10 мл разведенной соляной кислоты и далее поступают, как указано в статье «Нитритометрия» (стр. 799). В случае применения внутренних индикаторов используют нейтральный красный или тропеолин 00 в смеси с метиленовым синим. 1 мл 0,1 мол раствора нитрита натрия соответствует 0,01652 г C9H11NO2, которого в препарате должно быть не менее 99,5%. Хранение. Список Б. В хорошо укупоренной таре, предохраняющей от действия света. Высшая разовая доза внутрь 0,5 г. Высшая суточная доза внутрь 1,5 г. Местноанестезирующее средство. 2. Химическая схема синтеза Стадия 1. Получение п-нитробензойной кислоты. 1) 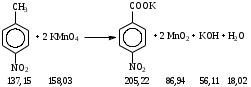 2) 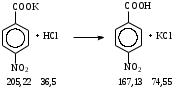 Стадия 2. Получение этилового эфира п-нитробензойной кислоты 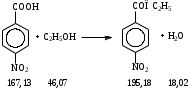 Стадия 3. Получение технического анестезина (восстановление «нитроэфира»). 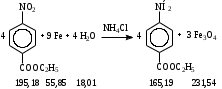 Роль хлористого аммония как электролита заключается в активации процесса влажной коррозии железа по схеме: 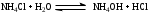 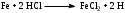 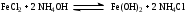 Приведенное выше суммарное уравнение процесса может быть выражено следующими частными уравнениями реакций восстановления: 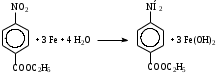 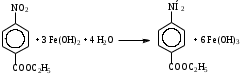 При осаждении ионов железа (в виде Fe(OH)2, Fe(OH)3, FeCO3, соответствующих оксидов железа (II и III)) кальцинированной содой имеют место также реакции нейтрализации хлороводорода и хлористого аммония: 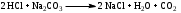 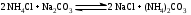 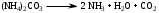 Возможная примесь непрореагировавшей исходной п-НБК после восстановления и обработки содой образует натриевую соль п-аминобензойной кислоты. 3. Экспериментальная часть Стадия 1. Получение п-нитробензойной кислоты. Загрузка: п-нитротоуол 13,7 г перманганат калия 39,5 г вода на промывку 43,2 г соляная кислота (35 %) 26 г В круглодонную колбу, снабженную мешалкой и термометром, загружают рассчитанное количество п-нитротолуола и 24-х кратное количество воды. Смесь нагревают на водяной бане и при температуре 83–85 °С вносят небольшими порциями в течение 3-х часов перманганат калия (2,5 моля на 1 моль п-нитротоуола). После прибавления всего перманганата калия температуру повышают до 98–100 °С и перемешивают 1 час. Затем реакционную массу в горячем виде фильтруют, осадок MnO2 на фильтре промывают горячей водой для извлечения адсорбированной соли п-нитробензойной кислоты. Фильтрат охлаждают до 5–10 °С, выдерживают 30 минут и фильтруют от остатков п-нитротоуола. Полученный раствор калиевой соли п-НБК подкисляют концентрированной соляной кислотой до полного осаждения п-НБК. При этом образуется суспензия с кремовым или слегка фиолетовым оттенком. Для обесцвечивания в суспензию добавляют на кончике скальпеля небольшое количество дитионита натрия (гидросульфита натрия) Na2S2O4. Далее суспензию отфильтровывают и промывают на фильтре водой (до отсутствия в осадке ионов Сl–). Осадок сушат при 70–80 °С или оставляют сушиться на воздухе до следующего занятия. Выход п-нитробензойной кислоты составляет 78 % от теоретического. п-Нитробензойная кислота представляет собой белые или слегка кремовые кристаллы, трудно растворимые в холодной воде, бензоле, хлороформе, сероуглероде, эфире. Тпл = 237–238 °С. Стадия 2. Получение этилового эфира п-нитробензойной кислоты. Загрузка: п-нитробензойная кислота 16,7 г этиловый спирт (96 %) 57 г серная кислота (93 %) 18 г сода кальцинированная (14 % раствор) до слабо-ще- лочной реак- ции на фе- нолфталеин В круглодонную колбу, снабженную мешалкой и обратным холодильником загружают этиловый спирт, серную кислоту, а затем прибавляют п-нитробензойную кислоту. При перемешивании подогревают реакционную массу в течение 6 часов при температуре кипения спирта. По окончании реакции охлаждают содержимое колбы, оставляют до следующего занятия. Выкристаллизовавшийся этиловый эфир п-нитробензойной кислоты со спиртовым маточником переносят в стакан и при наружном охлаждении размешивают стеклянной палочкой, нейтрализуют раствором кальцинированной соды (14 %) до слабощелочной реакции на фенолфталеин. После фильтрования осадок этилового эфира п-нитробензойной кислоты на воронке Бюхнера промывают водой и сушат при 40 °С. Выход от теоретического составляет 92 %. Этиловый эфир п-нитробензойной кислоты представляет собой белые кристаллы, плохо растворимые в холодной воде, хорошо в горячем этиловом спирте. Тпл = 56 °С. Стадия 3. Получение технического анестезина. Загрузка: «Нитроэфир» 13,3 г сухого Железный порошок 15,0 г Вода на восстановление 85,0 мл Аммоний хлористый (99,5 %) 1,5 г Сода кальцинированная (99,2 %) 1,0 г Изопропиловый спирт (87 %) 60–90 мл Уголь активированный 0,5 г Дитионит натрия (гидросульфит натрия) Na2S2O4 0,4 г Вода на промывку угля 5,0 мл Вода на промывку технического анестезина 50,0 мл В трехгорлую колбу, снабженную мешалкой, термометром и обратным холодильником, загружают 15,0 г железного порошка, 85,0 мл воды и 1,5 г хлористого аммония. Массу нагревают на кипящей водяной бане до 95–97 °С и начинают порциями добавлять измельченный «нитроэфир» (влажный или сухой), полученный на 1-ой стадии, поддерживая температуру в массе 98–102 °С. Общее время добавления «нитроэфира» – 20–40 минут. Затем дают выдержку при перемешивании на кипящей водяной бане в течение 1,5 часов. Реакционную массу охлаждают до 40–45 °С, добавляют кальцинированную соду, перемешивают. Далее массу охлаждают до 5–10 °С, отстаивают без перемешивания 10–15 минут, отсоединяют мешалку и раствор хлористого аммония и соды аккуратно сливают через фильтр, стараясь не взмучивать осадок в колбе (небольшой осадок шлама с фильтра возвращают в реакционную колбу). Вновь устанавливают мешалку и обратный холодильник и экстрагируют анестезин в 2–3 приема 87 % изопропиловым спиртом при температуре 65–70 °С при перемешивании по 10–15 минут (на одну экстракцию расходуется 20–30 мл спирта). После экстракции мешалку отсоединяют и горячий раствор сливают в коническую колбу1. Полученный раствор анестезина обрабатывают 0,5 г активированного угля и 0,4 г дитионита натрия (гидросульфита натрия), перемешивают 10–15 минут при 65–70 °С и отфильтровывают от угля в горячем виде. В фильтрате контролируют величину рН (5–5,5), при необходимости добавляют по каплям концентрированную соляную кислоту. Горячий раствор при перемешивании палочкой разбавляют небольшими порциями горячей воды (65–70 °С) до начала помутнения и кристаллизуют технический анестезин при температуре 5–7 °С на водяной бане или в холодильнике. Осадок отфильтровывают, тщательно промывают водой, отжимают на фильтровальной бумаге от влаги и передают на перекристаллизацию. Выход (по сухому продукту) составляет 97 % от теории, считая на «нитроэфир» Технический анестезин представляет собой белый кристаллический порошок с кремоватым оттенком. Содержание основного вещества 98,5–99,4 % от сухой массы. Стадия 4. Получение фармакопейного анестезина. Загрузки сырья на 1 г технического анестезина (с влагой до 10 %): На растворение: Изопропиловый спирт (50 %) 4,5 мл Уголь активированный 0,04 г Дитионит натрия (гидросульфит натрия) 0,007 г На промывку угля: Изопропиловый спирт (50 %) 1 мл На промывку фармакопейного анестезина: Изопропиловый спирт (50 %) 1,2 мл Дистиллированная вода 1,65 мл Технический анестезин растворяют в водном ИПСе при 75–80 °С, осветляют активированным углем и гидросульфитом натрия при этой температуре в течение 10–15 минут. Массу фильтруют в горячем виде через обогреваемую воронку, уголь промывают смесью ИПСа и воды. Объединенный фильтрат охлаждают в режиме самоохлаждения до 30–40 °С, затем на бане с солью и водой – до 0–5 °С и кристаллизуют анестезин в течение 1 часа. Анестезин отфильтровывают, промывают охлажденным до 0–5 °С водным изопропиловым спиртом, затем промывают дистиллированной водой и сушат при 40–50 °С. Выход составляет 89,5 % от теории (без учета регенерации маточников), считая на технический анестезин или 62,3 % от теории, считая на п-нитротолуол. Примечание: Вместо водного изопропилового спирта можно применять водный этиловый спирт той же концентрации. 1 |