Курсовая по теме наноинженерия. Качественный рентгеновский фазовый анализ с использованием систем ручного поиска
 Скачать 467.41 Kb. Скачать 467.41 Kb.
|

|
МИНИСТЕРСТВО НАУКИ И ВЫСШЕГО ОБРАЗОВАНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ Федеральное государственное бюджетное образовательное учреждение высшего образования «Ивановский государственный политехнический университет» (ИВГПУ)  Институт информационных технологий, естественных и гуманитарных наук Кафедра естественных наук и техносферной безопасности Курсовая работа На тему: «Качественный рентгеновский фазовый анализ с использованием систем ручного поиска» По дисциплине: «Рентгеновская дифрактометрия» Автор работы Уткин Д.Н. Подпись фамилия, инициалы Специальность 28.03.02 «Наноинженерия» Номер, наименование Номер зачетной книжки 195049 Группа «НИ-31» Руководитель работы к.т.н., доцент Комарова Т.А. Должность, подпись фамилия, инициалы Работа защищена___________ Оценка____________________ Иваново 2022 Содержание Введение................................................................................................................3 Глава 1. Обзор литературы..................................................................................4 1. Теория метода...................................................................................................4 2. Методика выполнения качественного фазового анализа.............................6 3. Приготовление объекта исследования............................................................8 4. Выбор режима съемки......................................................................................9 5. Выбор материала анода рентгеновской трубки............................................10 6. Расшифровка дифрактограммы......................................................................12 7. Описание рентгеновского дифрактометра D2 PHASER23..........................15 8. Подготовка образца к исследованию.............................................................16 Глава 2. Экспериментальная часть....................................................................18 9. Цели и задачи...................................................................................................18 10. Методическая часть.......................................................................................18 10.1 Характеристика объектов исследования и реактивов..............................18 11. Расчеты экспериментальной части..............................................................19 12. Заключение....................................................................................................22 13. Список используемой литературы..............................................................23 Введение В металлургической, химической, электронной и других отраслях промышленности, а также при выполнении научных исследований важно знать химический и фазовый состав материала, который является объектом производства или научно-исследовательской работы. Одним из современных и простых методов определения фазового состава кристаллических тел является рентгеновский. В основу метода положено явление дифракции рентгеновских лучей на кристаллической решетке. Каждая фаза имеет свою кристаллическую решетку. Под фазой понимают часть вещества, отделенную от других его частей границей раздела, при переходе через которую свойства меняются скачком. Для выполнения качественного и количественного фазового анализа используется современная рентгеновская аппаратура – рентгеновские дифрактометры. Она позволяет проводить его быстро и с большой точностью. В данной курсовой работе мы знакомимся с методом качественного фазового анализа. Глава 1. Обзор литературы 1.Теория метода Каждая твердая кристаллическая фаза имеет собственную, присущую ей кристаллическую решетку. Как правило, для сложных веществ фазовый состав отличается от их химического состава. Например, еcли мы имеем окисленную медь, то химический состав образца будет определяться процентным содержанием меди и кислорода. Фазовый же состав будет оцениваться весовым или молярным содержанием чистой меди и ее возможных оксидов CuO и Cu2O. При качественном фазовом анализе необходимо установить, какие фазы присутствуют в образце, а при количественном – найти их процентное содержание. Рентгеновский метод фазового анализа основан на том, что для рентгеновских лучей кристаллическая решетка является дифракционной. Условием дифракции рентгеновских лучей на кристаллической решетке является условие Вульфа-Брэгга: 2d sin θ = nλ, (1.1) где d – расстояние между соседними кристаллографическими плоскостями, с атомами которых взаимодействуют рентгеновские лучи; θ – угол под которым наблюдается дифракция; n – порядок дифракционного максимума (порядок «отражения»); λ – длина волны монохроматических рентгеновских лучей, падающих на кристалл. Если в качестве объекта использовать порошок или мелкокристаллический материал с различным образом ориентированными кристалликами, то при взаимодействии с ним монохроматических рентгеновских лучей всегда найдется для каждого сорта плоскостей определенное число кристалликов, попавших в «отражающее» положение. В этом случае под углом θ будет наблюдаться дифракционный максимум для данного сорта плоскостей. Угловое положение максимума будет определяться значением d, а последнее – геометрией кристаллической решетки. Интегральная интенсивность рефлекса IHKL, полученного от плоскостей с индексами(hkl) в n – ом порядке «отражения», причем H = nh, K= nk, L = nl, определяется выражением: I HKL = C ⋅ L( )θ ⋅  FHKL 2 ⋅ PHKL ⋅ e−2M ⋅ A(θ), (1.2) FHKL 2 ⋅ PHKL ⋅ e−2M ⋅ A(θ), (1.2)где C – общий для всех линий дифрактограммы множитель, зависящий от длины волны излучения; 2 FHKL – структурный фактор N 2πi(Hx j +Ky j +Lz j ) FHKL = ∑ f je , (1.3) j=1 fj – атомная амплитуда 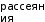 , зависящая от порядкового номера элемента; , зависящая от порядкового номера элемента; x, yj ,zj - координаты базисных атомов; PHKL – фактор повторяемости, учитывающий число эквивалентных плоскостей, дающих одну и ту же дифракционную линию. Он зависит от типа кристаллической решетки и сорта плоскостей;e-2M – температурный фактор; A(θ) – фактор поглощения, зависящий от исследуемого вещества, длины волны излучения и метода съемки. Интенсивность рефлекса зависит, кроме указанных выше факторов, от режима работы рентгеновского аппарата: тока через трубку; напряжения на трубке; размера щелей, режима работы счетчика квантов рентгеновского излучения, скорости вращения образца и счетчика, скорости протяжки диаграммной ленты. Наконец, интенсивность рефлекса определяется количеством данной фазы. Если исследуемый объект состоит из нескольких фаз, то каждой фазе будет соответствовать своя собственная дифракционная картина (рис. 1.1)  В этом случае дифрактограмма представляет собой наложение дифрактограмм всех имеющихся в исследуемом образце фаз. Интенсивность рефлексов каждой фазы будет зависеть от ее количества в исследуемой смеси. Так, из рис. 1.1 видно, что наиболее интенсивными являются рефлексы Cu, а самыми слабыми – Cu2O. Следовательно, в исследуемом образце меди содержится значительно больше, чем CuO и Cu2O. 2. Методика выполнения качественного фазового анализа Качественный фазовый анализ вещества можно проводить как в случае, когда предварительно известен химический состав и имеются данные о предполагаемом фазовом составе, так и в случае, когда об образце нет никаких сведений. Чаще всего приходится иметь дело с первой наиболее простой задачей. Об ее решении и будет идти речь ниже. Если известен химический состав и предполагаемый фазовый состав объекта анализа, то для его уточнения необходимо иметь литературные данные о значениях межплоскостных расстояний d/n и относительных интенсивностях рефлексов IHKL для каждой предполагаемой фазы. Такие сведения в настоящее время наиболее полно представлены в «Рентгенометрической картотеке», издаваемой до 1970 года Американским обществом по испытанию материалов (ASTM). Последующие выпуски издаются Объединенным комитетом порошковых дифракционных стандартов (JCPDS). Менее полными являются справочники, изданные на русском языке. [1], [2], [3]. Если данных о d/n и IHKL предполагаемой фазы в литературе нет, но есть сведения о структурном типе элементарной ячейки и ее параметрах, то нужно теоретически рассчитать дифрактограмму предполагаемой фазы. Если предполагаемая фаза имеется в чистом виде, то можно снять с нее дифрактограмму и экспериментально определить d/n и IHKL . Далее необходимо получить тем или иным экспериментальным методом дифрактограмму исследуемого объекта, а по ней определить d/n и IHKL всех дифракционных максимумов. При этом интенсивность всех рефлексов определяется по отношению к самому сильному, интенсивность которого принимается за 100. Начинают фазовый анализ с выяснения присутствия одной наиболее вероятной фазы. Для этого справочные данные о d/n и IHKL этой фазы сопоставляют с экспериментальными значениями d/n и IHKL, полученными в результате расчета дифрактограммы. Сопоставление начинают с самых ярких рефлексов (по справочным данным) с учетом возможной погрешности как экспериментальных значений d/n, так и справочных. Если данной фазы в исследуемой смеси мало, то ее даже самые сильные линии будут на суммарной дифрактограмме слабыми, а более слабые (по справочным данным) совсем не появятся. Проверив тщательно правильность отождествления рефлексов данной фазы (с учетом всех факторов, о которых будет идти речь дальше), тем же способом определяют наличие следующих предполагаемых фаз. В большинстве случаев для надежной идентификации фазы достаточно трех или четырех ее наиболее сильных рефлексов. Проведение фазового анализа образца, содержащего несколько фаз, осложняется тем обстоятельством, что рефлексы разных фаз могут накладываться, т.е. один и тот же рефлекс на дифрактограмме может принадлежать одновременно нескольким фазам. В этом случае для надежной идентификации фазы необходимо 7–10 рефлексов. Можно поступить и иначе: увеличить разрешающую способность дифрактограммы, что позволит разделить некоторые рефлексы (редко, когда d/n двух фаз точно равны). Далее, если данной фазы содержится очень мало, то ее рефлексы могут вообще не появиться на дифрактограмме или появятся один – два рефлекса. Чтобы убедиться в присутствии этой фазы, необходимо снять дифрактограмму при более высоких значениях общей интенсивности или часть дифрактограммы в тех углах, где находятся эти рефлексы. Фаза может дать слабые рефлексы не только из-за малого ее количества, но также из-за слишком больших размеров ее кристалликов (малое количество последних попадает в «отражающее» положение). В этом случае, чтобы убедиться в присутствии или, напротив, в отсутствии предполагаемой фазы, необходимо либо измельчить образец, либо снять дифрактограмму с вращением образца во время съемки. Мерой чувствительности рентгеновского фазового анализа служит минимальное количество вещества в смеси, дающее достаточный для определения наличия в образце комплект ее характерных рефлексов. Для разных фаз и разных смесей фаз чувствительность различна. Она тем больше, чем выше «отражательная» способность атомных плоскостей фазы, присутствие которой надо обнаружить, и чем слабее фон дифрактограммы. Существенно и соотношение коэффициентов поглощения всей смеси и определяемой фазы. Вещества, сильно рассеивающие рентгеновское излучение, легко обнаружить в слабо рассеивающей смеси. Соединения же легких элементов в смеси с соединениями тяжелых элементов можно обнаружить лишь при их больших содержаниях. В качестве иллюстрации сказанному выше рассмотрим несколько примеров. Смесь вольфрама (ОЦК решетка) с карбидом W2C (гексагональная решетка). Вольфрам можно обнаружить при содержании его 0,1– 0,2 вес. %, а W2C в случае, если его больше чем 0,3– 0,5 вес. %. Смесь металлических W (ОЦК решетка) и Cu (ГЦК решетка). Вольфрам обнаруживается при содержании 0,1 вес. %, а медь – при содержании 1 вес. %. Смесь Fe и его карбида Fe3C Fe3C можно обнаружить, если его содержится более 10 вес. % (он обладает сложной ромбической решеткой). В меди заметны 0,5 вес. % закиси меди Cu2O с кубической решеткой, а окись меди CuO с моноклинной решеткой дает заметные по интенсивности рефлексы при содержании ее в десять раз большем. Если линии определяемой фазы размыты из-за наличия микронапряжений, или дисперсности образца, или неоднородности его по составу, или, если имеем дело с твердым раствором, то чувствительность рентгеновского фазового анализа резко понижается. Чувствительность фазового анализа повышается, если фаза, которую надо выявить, имеет текстуру, т.е. кристаллики имеют преимущественную ориентацию какого-то определенного кристаллографического направления. Последнее приводит к тому, что некоторые линии на дифрактограмме усиливаются, а некоторые, наоборот, исчезают. Наличие сильных линий и позволяет выявить меньшее, чем в отсутствии текстуры, количество фазы. 3. Приготовление объекта исследования В дифрактометрии поликристаллов используется плоский образец. Обычно это порошок, тем или иным способом нанесенный на плоскую поверхность, либо таблетка, спрессованная из порошка, либо срез массивного поликристалла. Оптимальный размер частиц в образце должен быть порядка 10 мкм (10-5 см– 10-3 см). Один из способов приготовления образца для исследования таков. Стеклянную круглую пластинку, меньшую по диаметру 25 мм, слегка смазывают вазелином. На вазелин равномерным слоем насыпают порошок исследуемого материала (рис. 1.1, а). На порошок накладывают стеклянную пластинку и, слегка покачивая ее и постепенно увеличивая давление, разравнивают порошок и прессуют его (рис. 1.1, б). Полученный препарат крепят в стеклянной кювете на пластилине и придавливают сверху стеклянной пластинкой для того, чтобы поверхность образца оказалась параллельной краю кюветы (рис. 1.1, в). Кювету устанавливают в держатель гониометра и, если гониометр хорошо отюстирован, поверхность образца совмещается с плоскостью фокусировки. 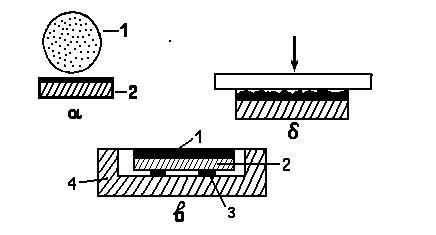 Рис. 1.2 4. Выбор режима съемки Основными параметрами съемки на дифрактометре являются: вещество анода и фильтра Кβ – излучения (или используемый монохроматор); величина высокого напряжения в киловольтах и ток через трубку в миллиамперах; тип счетчика; используемая шкала скорости счета импульсов; скорость движения счетчика в градусах в минуту и диаграммной ленты в миллиметрах в час; интервал между штрихами отметчика в градусах; размеры вертикальных щелей у трубки и счетчика в миллиметрах. Для определения фазового состава образца при съемке на дифрактометре необходимо подобрать такие ее условия, которые позволили бы при достаточно большой величине интенсивности рефлекса (для повышения чувствительности анализа) получить хорошую точность в определении углового положения рефлексов. Чтобы получить большую интенсивность, необходимо использовать широкие щели, большую постоянную времени, малую скорость движения счетчика. Напротив, точность в определении положения рефлекса увеличивается, если использовать узкие щели и малую постоянную времени. Экспрессность в проведении анализа требует значительной скорости движения счетчика. Режим съемки подбирается в лаборатории перед выполнением анализа или задается преподавателем. При правильно выбранном режиме съемки интенсивность самой сильной линии на дифрактограмме должна обеспечивать отклонение стрелки интенсиметра на всю выбранную шкалу. При этом, если рефлексы на дифрактограмме достаточно интенсивные, то лучше снимать дифрактограмму на шкале, соответствующей меньшей чувствительности интенсиметра. При выборе высокого напряжения руководствуются тем, что оно должно превышать в три-четыре раза минимальное напряжение, необходимое для установления тока насыщения. Последнее зависит от типа трубки, материала анода, тока накала трубки. 5. Выбор материала анода рентгеновской трубки Для съемки дифрактограммы надо правильно выбрать материал анода. При этом необходимо обеспечить следующие условия: Отсутствие вторичного характеристического излучения, вуалирующего дифрактограмму. Интенсивное вторичное излучение возникает в том случае, если атомный номер вещества анода на 2 – 3 единицы больше атомного номера элементов, входящих в состав исследуемого образца. Например, железо (Z =26), снимаемое на излучении трубки с медным анодом (Z =29), дает вторичное рентгеновское излучение, вуалирующее дифракционную картину. Наличие достаточного количества рентгеновских дифракционных максимумов. Достаточную разрешающую способность дифрактограммы. Первое правило не распространяется на те случаи, когда порядковый номер вещества анода намного выше порядкового номера элементов, содержащихся в объекте. В этом случае вторичное излучение можно легко отфильтровать. Чаще всего используются рентгеновские трубки с медным анодом. Из-за хорошей теплопроводности меди они выдерживают большие нагрузки. Излучение с более короткой длиной волны, чем λКαCu для фазового анализа используется редко. При съемке на коротковолновом излучении дифракционные линии «собраны» в малых углах и плохо разрешаются. Фазовый анализ требует использования монохроматического излучения. К – серия рентгеновского спектра, которая чаще используется для этих целей, состоит из двух основных линий: дублета Кα1α2 и Кβ . Излучение, соответствующее Кα1α2, примерно в пять раз сильнее излучения с длиной волны Кβ. Рентгеновские лучи с этой длиной волны мешают анализу, поэтому их отфильтровывают. Для этой цели достаточно поставить перед образцом фильтр – тонкую фольгу из вещества, содержащего элемент или состоящего целиком из элемента, порядковый номер которого на единицу, а для тяжелых анодов на две, меньше порядкового номера атомов вещества анода. Излучение с длиной волны Кα1α2 легко пройдет через этот фильтр. Лучи же с длиной волны Кβ выйдут из фильтра ослабленными во много раз. Это связано с тем, что при таком соотношении порядковых номеров вещества анода и фильтра скачок коэффициента поглощения µ лучей в веществе фильтра (рис. 1.3) лежит как раз между рефлексами, соответствующими длинам волн Кα1α2и Кβизлучения анода. Например, для излучения медного анода фильтром может служить никелевая фольга толщиной 0,007 мм. Несмотря на наличие фильтра Кβ рентгеновское излучение трубки не является строго монохроматичным. Наряду с Кα – излучением всегда присутствует излучение со сплошным спектром, дающее на дифрактограмме фон. Интенсивность фона наиболее велика в малых углах 2θ и уменьшается с углом 2θ до его значения, равного 90о. В больших углах фон от сплошного рентгеновского спектр может возрасти. Поскольку дифрактограму записывают в широком интервале углов, перо самописца и стрелка регистрирующего прибора могут уйти за пределы диаграммной ленты и шкалы. Чтобы этого не произошло, предварительно вручную выводят счетчик на минимум фона при 2θ, равном 90о (но не на дифракционный пик), устанавливают линию фона несколько выше нижнего края ленты и возвращают счетчик в исходное положение. 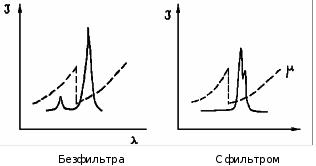 Рис. 1.3 Хорошую дифрактограмму (без фона и Кβ – рефлексов) можно получить, если использовать кристалл-монохроматор, который устанавливается либо между фокусом рентгеновской трубки и образцом (на первичном пучке), либо между образцом и счетчиком (на вторичном пучке). В качестве кристалловмонохроматоров чаще всего используются тонкие пластинки, вырезанные из монокристаллов кварца или графита так, что их поверхность параллельна кристаллографическим плоскостям кристалла с межплоскостным расстоянием dм. На рис. 1.2 приведена схема взаимного расположения фокуса рентгеновской трубки F, образца AOB, кристалла монохроматораCTD и счетчика C. Здесь О – ось вращения образца, Т – ось вращения кристалла-монохроматора, S1, S2 – щели. Кристалл – монохроматор устанавливается под углом θ к направлению рассеянных рентгеновких лучей, который удовлетворяет условию: 2dм sinθ=λ Kα , (1.4) 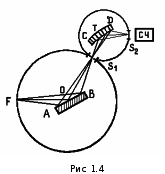 Рентгеновское излучение с другими длинами волн не попадает в щель счетчика. Поскольку использование кристалла для монохроматизации излучения сильно уменьшает нтенсивность рассеянных лучей обычно используется их фокусировка за счет изгиба кристалла. 6. Расшифровка дифрактограммы Дифрактограмма характеризуется положением и интенсивностью дифракционнвых максимумов. Положение пика измеряют углом отражения θ или 2θ, а интенсивность – высотой пика или площадью под ним. 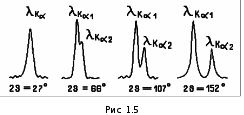 При измерении положения пиков, соответствующих длинам волн Кα1α2,возникает трудность за счет существования дублета. Дублет разрешается тем лучше, чем больше угол 2θ и меньше скорость вращения счетчика (рис. 1.5). В зависимости от степени разрешения дублета угловое положение дифракционного максимума измеряют в разных точках. При измерении положения пиков, соответствующих длинам волн Кα1α2,возникает трудность за счет существования дублета. Дублет разрешается тем лучше, чем больше угол 2θ и меньше скорость вращения счетчика (рис. 1.5). В зависимости от степени разрешения дублета угловое положение дифракционного максимума измеряют в разных точках. Из рис. 1.5 видно, что в малоугловой области (2θ < 45о) пики Кα1 и Кα2 сливаются настолько, что виден один практически симметричный пик с длиной волны λKα: λKα = λ Kα + λ Kα . (1.5) Его положение измеряют на высоте 1/2 от основания как среднее углов, отвечающих точкам с одинаковой интенсивностью. При больших углах «отражения» (2θ ≈40 –60о) с правой стороны пика появляется «платформа» – компонента Кα2, но разрешение пика еще недостаточно для разделения пиков λКα1 и λКα2. И в этом случае указанным выше способом измеряют значение угла 2θ, соответствующее λKα. При дальнейшем увеличении угла «отражения» (2θ =60 –80о) появляется возможность измерения положения пиков, соответствующих λКα1 и λКα2. В этом случае более точные результаты дает интенсивный пик λКα1. Если на дифрактограмме присутствует сильный фон, интенсивность которого изменяется с углом (рис. 1.6), и пик уширен, то для нахождения угла 2θ, отвечающего его максимуму, соединяют точки одинаковой интенсивности (отсчитанной от уровня фона) на расстоянии от его вершины не большем 1/3 высоты пика. Линии, соединяющие эти точки, проводят параллельно линии фона АВ. Через середины отрезков этих линий, лежащих внутри профиля, проводят прямую, пересечение которой с профилем линии (точка С на рис. 1.6) и определяет угловое положение дифракционного пика (2θэксп, рис. 1.6). 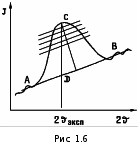 Для определения численного значения углов при записи дифрактограммы на диаграммную ленту с помощью отметчика углов на ней ставятся метки через градус или 0,1 градуса (рис. 1.6). Угол 2θэксп определяют измеряя линейкой (лучше с ценой деления 0,5 мм) расстояние до ближайшей метки. Суммарная погрешность в определении углового положения максимума рефлекса 2θэксп складывается из ошибки в определении точки, соответствующей максимуму на шкале углов, ошибки определения расстояния до ближайшей метки и систематической погрешности, обусловленной неточностью установки нуля шкалы отсчета углов на дифрактометре. Источниками систематической погрешности являются: неточная юстировка прибора, (неточная установка нулевого положения счетчика, смещение плоскости образца с оси гониометра, непараллельность оси гониометра и выходных щелей), проникновение рентгеновских лучей вглубь образца, вертикальная расходимость первичного и дифрагированных пучков, преломление рентгеновских лучей, изменение спектрального распределения лучей при прохождении их через фильтр и др. Исключить эти ошибки можно проводя съемку со стандартом. Стандарт должен иметь достаточно точно известные параметры кристаллической решетки или значения d/n. Поэтому в качестве стандарта лучше использовать чистые вещества с кубической решеткой, обладающие высокой рассеивающей способностью. К таким веществам относятся, например, W, Ge, Si, Pt, кварц, двуокись циркония. Стандартное вещество лучше подмешивать к исследуемому материалу, если он представляет собой порошок, что позволяет исключить все источники систематической погрешности. Но для фазового анализа можно использовать внешний стандарт, сняв с него дифрактограмму перед выполнением работы по определению фазового состава исследуемого материала. При этом будут исключены основные систематические погрешности. Численное значение систематических погрешностей для используемого диапазона углов 2θ может быть найдено, если снять дифрактограмму эталона, рассчитать по ней значения углов, соответствующих его рефлексам, сравнить эти значения с табличными для данного вещества эталона, найти разности углов ∆2θсист=(2θэксп–2θтабл) и построить график зависимости ∆2θсист от 2θ. Пользуясь этим графиком можно найти систематическую погрешность для 2θэксп исследуемого образца и устранить её. По исправленным на систематическую погрешность значениям углов дифракции всех рефлексов на дифрактограмме θэксп необходимо по формуле (1.1) рассчитать величины d/n и случайные погрешности их определения: ⎛ d ⎞ d  ∆⎜ ⎟ = ctgθ ⋅ ∆θ (1.6) ⎝ n ⎠ n Для качественного анализа одних значений d/n недостаточно. Необходимы, как указывалось в разделе 1.2, значения относительных интенсивностей рефлексов. Если рефлексы узкие, то можно измерять интенсивность по их высоте в произвольных единицах. При этом перед измерениями высоты рефлексов на дифрактограмме проводят плавную кривую линии фона, от которой и ведут измерения. Если рефлексы широкие, то определяют их интегральную интенсивность, измеряя площадь под линией дифракционного пика или используя на дифрактометре режим съемки интегральной интенсивности. 7. Описание рентгеновского дифрактометра D2 PHASER23 Дифрактометр рентгеновский D2 PHASER предназначен для измерения параметров кристаллической решетки методом порошковой дифрактометрии. Принцип действия дифрактометра основан на дифракции рентгеновских лучей от атомных плоскостей кристаллической решетки исследуемого вещества. Дифракция рентгеновских лучей соответствует закону ВульфаБрегга. Конструктивно дифрактометра состоит из источника рентгеновского излучения с анодами из меди, кобальта, хрома, молибдена, железа, вольфрама, титана или серебра, гониометра, блоков детектирования и системы управления, сбора и обработки данных. Дифрактометр построен по оптической схеме Брегга-Бретанно, в которой плоский образец пробы находится в центре гониометра. Дифрактометр выполнен в виде единого модуля, внутри которого расположены все составляющие элементы, включая управляющий компьютер и замкнутый цикл охлаждения рентгеновской трубки. Регистрация дифракционной картины осуществляется при синхронном повороте блока детектирования и рентгеновской трубки вокруг общей оси гоноиметра с требуемыми угловыми скоростями. Для обеспечения высокой точности отсчета угла в дифрактометре используется специальные оптические кодовые датчики. В дифрактометрах для регистрации квантов рентгеновского излучения устанавливается позиционно-чувствительный или сцинтилляционный детектор. Фотография общего вида дифрактометра приведена на рисунке 7.  Рис.7. Рентгеновский дифрактометр D2 PHASER23 Дифрактометр применяется для контроля производства и качества продукции в металлургической, горнодобывающей, керамической, целлюлозно-бумажной, фармацевтической промышленностях, а также для проведения научных исследований. В дифрактометре применяется программное обеспечение, которое позволяет выполнять настройку параметров измерений, производить сбор, обработку и хранение данных. Макс. угловой диапазон: 3 — 140 ° 2Theta (в зависимости от детектора) (140, т.к. линейный детектор LYNXEYE). Точность определения: ± 0.02° во всем измерительном диапазоне. 8. Подготовка образца к исследованию Для получения достаточно хороших рентгенограмм необходимо тщательно готовить образцы для съемки. Самым распространенным методом съемки является метод порошка, по которому образец является поликристаллическим телом, полученным из тонко измельченного порошка. Для съемки на фотопленке порошок наносится на нить или набивается в капилляр диаметром 0,7 мм. Для съемки дифрактограмм (рентгенограмм на самописец) порошок насыпается и фиксируется в углублении специальной кюветы из кварцевого стекла, если для фиксации недостаточно простого придавливания, применяют органические клеи (БФ, цапонлак и т.д., а также обычные технические масла). Кроме того, порошок можно спрессовать в виде таблетки диаметром до 25 мм или применить для съемки плоский образец в твердом виде. Важным фактором, определяющим чувствительность метода, является размер кристалликов исследуемого вещества. Поэтому следует обратить внимание на тщательность растирания порошка, так как порошок, состоящий из крупных кристаллов, дает нечеткие, малоинтенсивные рентгенограммы. Растирать порошок следует в агатовой ступке агатовым пестиком (для исключения загрязнения пробы) до прохождения через сито 10000 отв./см² (0,000063; 63 мкм). Оптимальный размер кристаллов около 5 - 10 мкм. Вместе с тем в некоторых случаях нельзя сильно перетирать пробу, так как сильное воздействие (особенно с давлением) приводит к нарушению кристаллической структуры препарата, появлению напряжений в кристаллах (а значит, к ухудшению качества рентгенограммы). В некоторых случаях при растирании пробы происходит полиморфное превращение. Это явление можно использовать при исследовании стабильности материалов. Но, следует помнить также, что в случае, если размер кристалликов ниже определенного (менее 0,1мкм), то интерференционные линии могут быть размыты; и при малом количестве фазы её линии могут сливаться с фоном. Керамика, ситалл или изделия на основе вяжущих материалов чаще всего представляют собой поликристаллический материал. Поэтому при фазовом анализе можно использовать непосредственно поверхность образца или воспользоваться методом порошка. Глава 2. Экспериментальная часть 9. Цели и задачи Провести качественный рентгенофазовый фазовый анализ с помощью полученных дифрактограмм на рентгеновском аппарате D2 PHASER23. 10. Методическая часть 10.1. Характеристика объектов исследования и реактивов Объекты исследования: -Размельченный CaCO ( школьный мел)  Рис.8. Порошок CaCO Оборудование: - рентгеновский дифрактометр D2 PHASER23 Технические характеристики рентгеновского дифрактометра D2 PHASER23 В дифрактометре (рис.7) применяется программное обеспечение, которое позволяет выполнять настройку параметров измерений, производить сбор, обработку и хранение данных. Программное обеспечение: • Совместимость со всей линейкой рентгеновских систем Bruker AXS • Работа в сети • Поддержка многопользовательского режима • Программа EVA для фазового анализа • Программа TOPAS для количественного фазового и структурного анализа Основные технические характеристики: • Материал анода: стандартная отпаянная рентгеновская трубка: CuKα и CoKα; • Размер фокуса - 0.4 x 12 мм; • Номинальный режим работы источника рентгеновского излучения: 30 кВ /10 мА; • Вертикальный Theta / Theta гониометр, радиус 140 мм; • Метод сканирования θs/θd связанные; • Диапазон углов сканирования 2θ: от -3 до 160°; • Минимальный шаг сканирования 0.01°; • Детектор отраженных рентгеновских лучей: твердотельный позиционно-чувствительный детектор LYNXEYE: o количество каналов регистрации 190; o эффективность регистрации рентгеновского Cu-K(aльфа)- излучения не менее 98%; o пространственное разрешение детектора 75 мкм. • Оптическая система: o щели на выходном пучке фиксированные; o щели на дифрагированном пучке фиксированные. 11. Расчеты экспериментальной части На рисунке 9 предоставлена, полученная дифрактограмма CaCO , по которой мы будем проводить рентгенофазовый анализ. Рис.9. Полученая дифрактограмма CaCO 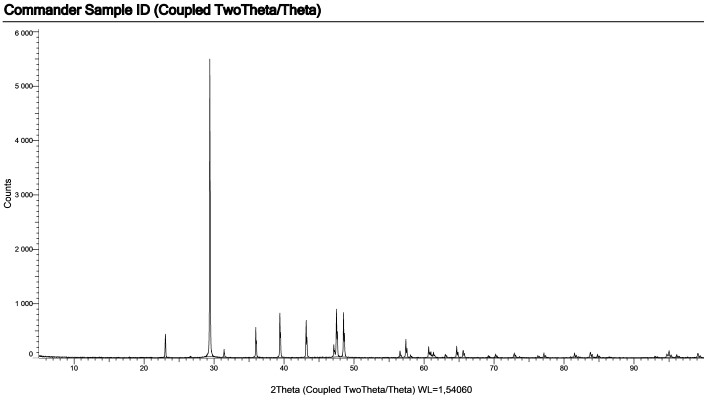 Для дальнейшего анализа возьмем 5 первых максимальных пиков (рис.10).  Рис. 10. Дифрактограмма CaCO , 5 первых пиков Табл.1 Расчет по дифрактограмме
По формуле: для 5 первых пиков. В таблице 1, занесены межплоскостные расстояния углов и их интенсивность. Как мы видим на дифрактограмме CaCO₃ (рис.10), аморфной области нет, но при этом присутствует лишь кристаллическая. Из этого можно сделать вывод, что степень кристалличности будет P=100%. 12. Заключение В ходе работы с помощью рентгеноструктурного анализа на рентгеновском дифрактометре D2 PHASER23 мы изучили качественный метод. Качественный фазовый анализ проводят сравнением экспериментальных значений межплоскостных расстояний и относительных интенсивностей с эталонными рентгенограммами, так как каждое вещество имеет свою «картину» расположения линий на рентгенограмме. Качественный фазовый анализ позволяет разделять и идентифицировать отдельные фазы гетерогенной системы. Объектами исследования в фазовом анализе являются металлы, сплавы, химические соединения, минералы, руды. С помощью рентгенофазового анализа можно определить состав неметаллических включений в металлах (оксидов, сульфидов, нитридов, карбидов), распределение легирующих элементов в многофазных сплавах. Широкое применение рентгенофазового анализа объясняется хорошо разработанной теорией, простотой приготовления образцов, относительной экспрессностью получения качественных результатов, сохранением образцов без изменения после исследования, возможностью использования поликристаллического материала, возможностью массовых измерений, возможностьюразличения полиморфных модификаций, возможностью получения из экспериментальной дифрактограммы, наряду с данными о фазовом составе, данных о структурных характеристиках отдельных фаз и их количестве. 13. Список используемой литературы 1. Горелик, С.С. Рентгенографический и электронноптический анализ/. С.С. Горелик, Л.Н. Расторгуев, Ю.А Скаков; М. : Металлургия, 1970. 368 с. 2. Китайгородский, А.И. Рентгеноструктурный анализ мелкокристаллических и аморфных тел; М. : Изд. техн.-теор. л-ры, 1952. 588 с. 3. Миркин, Л.Н. Справочник по рентгеноструктурному анализу поликристаллов; М. : Гос. изд. физ.-мат. л-ры, 1961. 863 с. 4. Гуревич, А.Г. Физика твердых тел.- Учеб. пособие для вузов / ФТИ им. А.Ф. Иоффе РАН.- СПб.: Невский Диалект; БВХ-Петербург, 2004.-320 с.: ил. 5. Жданов, Г.С. Основы рентгеноструктурного анализа.- Москва.- Гостехиздат.-1940.-76 с.: ил. 6. Покоев, А.В. Рентгеноструктурный анализ.- Москва.- изд. 2,- 1981.- 127 с. 7. Рахимова, Н.Т. Курсовая на тему "Рентгеноструктурный анализ".- Уфа.-2012.-30 с. 8. Белов, Н.В. Структурная кристаллография.- Санкт-Петербург.- изд. 4, 1951.-97 с. 9. "Wikipedia".- Интернет-энциклопедия 10. Джеймс, Р. Оптические принципы дифракции рентгеновских лучейМосква.- Гостехиздат.-изд.1, 1950.-146 с.: ил. 11. Johnston W.D., Jr. Nonlinear optical coefficients and the Raman scattering efficiency of LO and TO phonons in acentric insulating crystals // Phys. Rev. B. - 1970. - V.1, №8. - P.3494-3503. 12. Л. М. КОВБА, В. К. ТРУНОВ, Ренгенофазовый анализ, Издание второе, дополненное и переработанное, издательство московкого университета 1976 13.https://www.melytec.ru/upload/iblock/5b6/Порошковый%20дифрактом етр%20D2%20PHASER_rus.pdf |