Катализатор каталитического крекинга и каталитического риформинг. Катализатор каталитического крекинга и каталитического риформинга
 Скачать 257.56 Kb. Скачать 257.56 Kb.
|

|
«Катализатор каталитического крекинга и каталитического риформинга» Содержание 1 Описание процессов Каталитический крекинг Каталический риформинг 2. Катализаторы 2.1 Катализаторы каталитического крекинга 2.1.1 Строение катализаторов каталитического крекинга 2.1.2 Механизм процессов на катализаторе 2.1.3 Промышленный марки катализаторов каталитического крекинга 2.2 Катализаторы каталитического риформинга 2.2.1 Состав катализаторов каталитического риформинга и механизм 2.2.2 Механизм и химизм процесса каталитического риформинга 2.2.3 Марки катализаторов каталитического риформинга 1 Описание процессов 1.1 Каталитический крекинг Назначение: производство с максимально высоким выходом (до 50 % и более) высокооктанового бензина и ценных сжиженных газов Сырье: вакуумный дистиллят (газойль) широкого фракционного состава (350…500 °С). В ряде случаев в сырье крекинга вовлекаются газойлевые фракции термодеструктивных процессов, гидрокрекинга, рафинаты процессов деасфальтизации мазутов и гудронов, полупродукты масляного производства и др На современных зарубежных установках перешли к переработке глубоковакуумных газойлей с температурой конца кипения 540…620 °С. На специально запроектированных установках каталитическому крекингу подвергают остаточное сырье: мазуты и даже гудроны или их смеси с дистиллятным сырьем 643 без или после предварительного облагораживания гидроочисткой, деасфальтизацией или деметаллизацией. Продукты: - сжиженный газ - сырья для последующих производств высокооктановых компонентов бензинов изомерного строения: алкилата и метил-трет-бутилового эфира, а также сырья для нефтехимических производств; - высокооктановый бензин; - легкий газойль используется обычно как компонент дизельного топлива; -тяжелый газойль с высоким содержанием полициклических ароматических углеводородов — как сырье для производства технического углерода или высококачественного электродного кокса (например, игольчатого); - кокс. Оптимальные технологические параметры каталитического крекинга зависят от выбранной схемы процесса. В большинстве технологических схем температура в реакторе составляет порядка 510-540 0С, давление поддерживается практически постоянным и составляет 0,5-2 атм. Повышение давления несколько ухудшает селективность процесса и приводит к росту газо- и коксообразования. 1.2 Каталический риформинг Назначение: повышения детонационной стойкости бензинов и получения индивидуальных ароматических углеводородов, главным образом бензола, толуола, ксилолов — сырья нефтехимии. Важное значение имеет получение в процессе дешевого водородсодержащего газа для использования в других гидрокаталитических процессах. Сырье: прямогонные бензиновые фракции, бензины вторичных процессов - гидрокрекинга, термического крекинга и т.д., при условии их специальной подготовки. При получении высокооктанового компонента автомобильного бензина используются широкие фракции, выкипающие в пределах от 60-90°С до 180°С; при получении бензола, толуола, ксилолов – узкие фракции, выкипающие соответственно в интервалах 62-85°С, 85-105°С, 105-140°С. Для предотвращения дезактивации катализатора в сырье ограничивается содержание серы (не более 0,00005÷0,0010 % в зависимости от типа катализатора) и азота (не более 0,0001%). Продукты: - компонент высокооктанового бензина - узкие фракции, являющиеся сырьем установок экстракции ароматических углеводоров - Водородсодержащий газ, используется в процессах гидроочистки, изомеризации, гидрокрекинга - углеводородный газ - головка стабилизации, являющаяся сырьем установок ГФУ Технологические параметры. Основными технологическими параметрами процесса каталитического риформинга являются: температура, давление, объёмная скорость подачи сырья, мольное соотношение водород: углеводороды (кратность циркуляции водородсодержащего газа), мольное соотношение вода: хлористый водород, определяющее содержание хлора на катализаторе. Температура является основным регулируемым параметром процесса. Процесс риформирования проводят в реакторе в интервале температур 480—530 °С.Увеличение температуры процесса приводит к увеличению глубины ароматизации парафиновых углеводородов. Снижение температуры ниже 480 °С приводит к увеличению селективности, что объясняется изменением соотношения скоростей реакций ароматизации и гидрокрекинга в пользу первых. Однако работа в области высоких температур, обеспечивающих более высокие глубины и селективность ароматизации парафиновых углеводородов, затруднена высокой скоростью дезактивации катализатора вследствие его закоксовывания. Давление является вторым по значимости технологическим параметром процесса каталитического риформинга. Снижение давления приводит к увеличению селективности процесса риформинга.с увеличением выхода жидкого продукта и водорода, уменьшением выхода лёгких углеводородов С1-С4. Вместе с тем, снижение давления приводит к увеличению скорости дезактивации катализатора, уменьшению межрегенерационного периода, особенно в области давлений ниже 1,5 МПа. Увеличение объёмной скорости подачи сырья приводит к увеличению выхода жидкого продукта при одновременном снижении выхода ароматических углеводородов. Мольное соотношение водород / углеводороды, характеризуемое в промышленной практике кратностью циркуляции водородсодержащего газа, оказывает существенное влияние на стабильность работы катализатора риформинга. 2. Катализаторы 2.1 Катализаторы каталитического крекинга 2.1.1 Строение катализаторов каталитического крекинга Промышленные катализаторы крекинга представляют собой в этой связи сложные многокомпонентные системы, состоящие: 1) из матрицы (носителя); 2) активного компонента — цеолита; 3) вспомогательных активных и неактивных добавок. Матрица катализаторов крекинга выполняет функции как носителя — поверхности, на которой затем диспергируют основной активный компонент — цеолит и вспомогательные добавки, так и слабого кислотного катализатора предварительного (первичного) крекирования высокомолекулярного исходного нефтяного сырья. В качестве материала матрицы современных катализаторов крекинга преимущественно применяют синтетический аморфный алюмосиликат с высокой удельной поверхностью и оптимальной поровой структурой, обеспечивающей доступ для крупных молекул крекируемого сырья. Аморфные алюмосиликаты являлись основными промышленными катализаторами крекинга до разработки цеолитсодержащих катализаторов. Синтезируются они при взаимодействии растворов, содержащих оксиды алюминия и кремния, например жидкого стекла Na2O · 3SiO2 и сернокислого алюминия Al2(SO4)3. Химический состав аморфного алюмосиликата может быть выражен формулой Na2O(Al2O3 · xSiО2) где х — число молей SiO2 на 1 моль Al2O3. Обычно в промышленных аморфных алюмосиликатах содержание оксида алюминия находится в пределах 6…30 % мас. Аморфные алюмосиликаты обладают ионообменными свойствами, а для придания каталитической активности обрабатывают их раствором сернокислого алюминия для замещения катионов Na+ на Аl3+. Высушенные и прокаленные аморфные алюмосиликаты проявляют протонную и апротонную кислотности. При этом по мере повышения температуры прокаливания происходит превращение протонных кислотных центров в апротонные. Активным компонентом катализаторов крекинга является цеолит,который позволяет осуществлять вторичные каталитические превращения углеводородов сырья с образованием конечных целевых продуктов. Цеолиты (от греч. цео — кипящий, литос — камень) представляют собой алюмосиликаты с трехмерной кристаллической структурой следующей общей формулы: Me2/nO · Аl2О3 · xSiO2 · уН2О , где n — валентность катиона металла Me; х — мольное соотношение оксидов кремния и алюминия, называемое силикатным модулем; у — число молей воды. В настоящее время насчитывается несколько десятков разновидностей природных и синтетических цеолитов, отличающихся структурой, типом катионов Me, силикатным модулем и числом молекул кристаллизационной воды. Структура цеолитов характеризуется наличием большого числа полостей, соединенных между собой окнами, или микроканалами, размеры которых сравнимы с размерами реагирующих молекул. Обычно полости имеют больший диаметр, чем каналы (или окна). Например, в цеолите типа шабазит имеется 3 · 1020 полостей диаметром 11,4 Å, в каждую полость которого может поместиться 24 молекулы воды. Диаметр окон шабазита составляет 4,9 Å. При нагреве цеолита вода удаляется и образуется ячеистая структура. Удельная поверхность цеолитов достигает 700…1000 м/г. Обезвоженные цеолиты способны избирательно адсорбировать молекулы различных веществ в зависимости от размеров каналов. Разумеется, если диаметр адсорбируемого вещества больше, чем сечение канала, то оно не может проникнуть во внутренние поры цеолита (ситовой эффект). Так, при диаметре канала (окна) 4 Å цеолит не может адсорбировать углеводороды нормального строения, диаметр молекул которых равен = 4,9 Å. Обычно тип структуры синтетического цеолита обозначают буквами латинского алфавита А, X, Y, ... L и т. д. Перед буквами ставят химическую формулу катиона металла, компенсирующего отрицательный заряд алюминия в алюмосиликате. Например, СаХ означает цеолит типа X в кальциевой обменной форме; LaY, ReY — соответственно лантановая и редкоземельная форма цеолита Y. Цеолиты типа А, имеющие малые размеры окон (3,3…5 Å) и небольшой силикатный модуль (1,8…2,0), как правило, не используются в каталитических процессах и применяются в качестве адсорбентов. В каталитических процессах, в том числе крекинга нефтяного сырья, наибольшее применение нашли цеолиты типа X и Y — оба аналоги природного фожазита. В последние годы широкое распространение получают высококремнеземные трубчатые цеолиты L с силикатным модулем более 30 (например, ZSM). 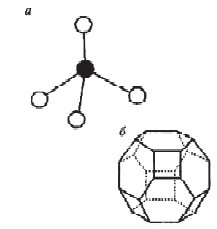 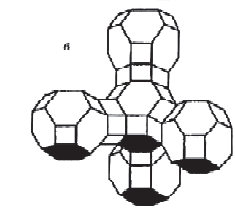 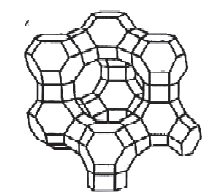 а — тетраэдр; б — содалитовая клетка; в — суперклетка; г — элементарная ячейка Рисунок 1- Строение цеолитов типа фожазита Первичной основой (структурной единицей) кристаллической решетки цеолитов X и Y является тетраэдр, состоящий из четырех анионов кислорода, которые окружены значительно меньшими по размерам ионами кремния или алюминия (рисунок 1). 24 тетраэдра образуют вторичную структурную единицу —усеченный октаэдр (кубооктаэдр, который содержит восемь шестиугольных и шесть квадратных поверхностей), так называемую содалитовую клетку. На следующей ступени структурирования четыре кубооктаэдра объединяются в тетраэдрическую конфигурацию вокруг пятого при помощи шестиугольных призм, образуя суперклетку (рисунок 1). В результате объединения множества суперклеток (в фожазите их восемь) в регулярную систему формируется элементарная ячейка цеолита (рисунок 1). Тетраэдры из оксидов кремния и алюминия расположены так, что цеолиты имеют открытые участки структуры. Это и создает систему пор с высокой удельной поверхностью. Химическую формулу первичной структурной единицы — тетраэдров кремния и алюминия — можно представить в виде: 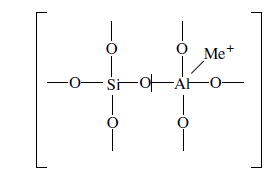 Тетраэдры с ионами Si4+ электрически нейтральны, а тетраэдры с ионами трехвалентного алюминия Аl3+ имеют заряд минус единица, который нейтрализуется положительным зарядом катиона Ме+ (сначала катионом Na+, поскольку синтез чаще ведется в щелочной среде, затем в результате катионного обмена — катионами других металлов, катионом NH4 или протоном Н+). Наличие заряженных ионов алюминия на поверхности цеолита (центры Бренстеда) и обусловливает кислотные свойства и, следовательно, его каталитическую активность. Натриевая форма цеолитов каталитически малоактивна и наименее термостабильна. Оба эти показателя существенно улучшаются при увеличении силикатного модуля цеолитов, а также степени ионного обмена на двухвалентные и особенно на трехвалентные металлы. Среди них более термостабильны цеолиты типа ReY, обладающие к тому же важным свойством — высокой каталитической активностью. Благодаря этим достоинствам цеолиты серии ReY как активный компонент катализаторов крекинга получили исключительно широкое применение в мировой нефтепереработке. Важным этапом в области дальнейшего совершенствования цеолитных катализаторов крекинга явилась разработка (в 1985 г. Фирмой 654 «Юнион карбаид») нового поколения цеолитов, не содержащих редкоземельных элементов, — так называемых химически стабилизированных цеолитов. В условиях воздействия высоких температур и водяного пара цеолиты ReY даже при полном редкоземельном обмене подвергаются частичной деалюминации: 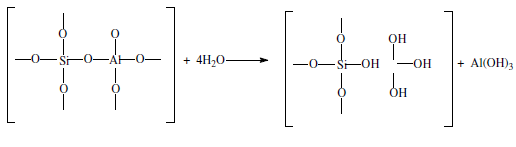 В результате гидродеалюминации в суперклетке образуется пустота, что является причиной постепенного разрушения кристалла цеолита. Гидроксид алюминия, который не выводится из кристалла, а откладывается внутри суперклетки цеолита, обладает, кроме того, нежелательной каталитической активностью (кислотностью Льюиса, ускоряющей реакции образования легких газов и кокса). Химическая стабилизация цеолитов заключается в низкотемпературной химической обработке их фторосиликатом аммония по реакции: 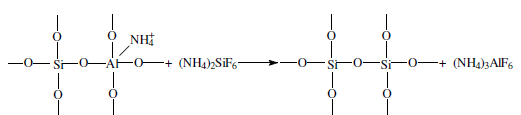 В результате обмена ионов Аl на ионы Si образуется более прочный и термостабильный цеолит с повышенным силикатным модулем и кристаллической решеткой без пустот. Еще одно достоинство этого процесса, обозначенного как процесс LS-210, — это то, что фтороалюминат аммония растворим и полностью выводится из кристаллической решетки цеолита. Цеолиты LS-210 (торговые марки Альфа, Бета, Эпсилон и Омега) характеризуются повышенной гидротермической стабильностью и селективностью, повышенной стабильностью по отношению к дезактивации металлами, но пониженной активностью в реакциях переноса водорода, что способствует повышению выхода изоолефинов в газах крекинга и октановых чисел бензинов. Недостатком всех цеолитов является их не очень высокая механическая прочность в чистом виде, и потому они в качестве промышленного катализатора не используются. Обычно их вводят в диспергированном виде в матрицу катализаторов в количестве 10…20 % мас. Вспомогательные добавки улучшают или придают некоторые специфические физико-химические и механические свойства цеолитсодержащим алюмосиликатным катализаторам (ЦСК) крекинга. ЦСК без вспомогательных добавок не могут полностью удовлетворять всему комплексу требований, предъявляемых к современным промышленным катализаторам крекинга. Так, матрица и активный компонент — цеолит, входящий в состав ЦСК, обладают только кислотной активностью, в то время как для организации интенсивной регенерации закоксованного катализатора требуется наличие металлических центров, катализирующих реакции окислительно-восстановительного типа. Современные и перспективные процессы каталитического крекинга требуют улучшения и оптимизации дополнительно таких свойств ЦСК, как износостойкость, механическая прочность, текучесть, стойкость к отравляющему воздействию металлов сырья и т. д., а также тех свойств, которые обеспечивают экологическую чистоту газовых выбросов в атмосферу. Ниже приводится перечень наиболее типичных вспомогательных добавок: а) в качестве промоторов, интенсифицирующих регенерацию закоксованного катализатора, применяют чаще всего платину, нанесенную в малых концентрациях (< 0,1 % мас.) непосредственно на ЦСК или на окись алюминия с использованием как самостоятельной добавки к ЦСК. Применение промоторов окисления на основе Pt позволяет значительно повысить полноту и скорость сгорания кокса катализатораи, что не менее важно, существенно понизить содержание монооксида углерода в газах регенерации, тем самым предотвратить неконтролируемое загорание СО над слоем катализатора в регенераторе, приводящее к прогару циклонов, котлов-утилизаторов и другого оборудования(из отечественных промоторов окисления можно отметить КО-4, КО-9, Оксипром-1 и Оксипром-2); б) с целью улучшения качества целевых продуктов в последние годы стали применять добавки на основе ZSM-5, повышающие октановое число бензинов на 1…2 пункта; в) для снижения дезактивирующего влияния примесей сырья наЦСК в последние годы весьма эффективно применяют технологию каталитического крекинга с подачей в сырье специальных пассиваторов металлов, представляющих собой металлоорганические комплексы сурьмы, висмута, фосфора или олова. Сущность эффекта пассивации заключается в переводе металлов, осадившихся на катализаторе, в неактивное (пассивное) состояние, например, в результате образования соединения типа шпинели. Пассивирующий агент вводят в сырье в виде водо- или маслорастворимой добавки. Подача пассиваторов резко снижает выход кокса и водорода, увеличивает выход бензина и производительность установки (в настоящее время пассиваторы применяют на 80 % установок каталитического крекинга остатков в США и около 50 % установок в Западной Европе). В последние годы внедряется ЦСК с твердой добавкой — ловушкой ванадия и никеля, содержащей оксиды Са, Mg, титанат бария и др., адсорбирующие в 6...10 раз больше металлов, чем сам катализатор; г) при каталитическом крекинге негидроочищенного сырья образуются (в регенераторе) оксиды серы и азота, отравляющие атмосферу. В связи с возросшими требованиями к экологической безопасности промышленных процессов исключительно актуальной становится проблема улавливания вредных компонентов газовых выбросов. Если в состав ЦСК ввести твердую добавку MgO или СаО, то такой катализатор становится переносчиком оксидов серы из регенератора в реактор по схеме: в регенераторе: MgO + SO3 → MgSO4 ; в реакторе: MgSO4 + 4Н2 → MgO + H2S + 3H2O ; или 2MgSO4 + СН4 → 2MgO + 2H2S + СО2 . Образующийся сероводород, выводимый из реактора вместе с продуктами крекинга, будет извлекаться затем из газов аминной очисткой; д) для повышения механической прочности ЦСК в состав аморфной матрицы дополнительно вводят тонкодисперсную окись алюминия (α-форму). Кроме того, для снижения потерь катализатора от испарения и уменьшения коррозии аппаратуры в системах катализатора в циркулирующий катализатор вводят смазывающие порошки из смеси окиси магния, карбоната и фосфата кальция, иногда титаната бария. Эти добавки взаимодействуют при высокой температуре с поверхностью катализатора, в результате чего на ней образуется глянец, способствующий снижению истирания. 2.1.2 Механизм процессов на катализаторе Химические превращения углеводородов крекируемого сырья, протекающие по карбений-ионному цепному механизму на поверхности ЦСК, можно представить в целом в следующей последовательности. 1. Первичные мономолекулярные реакции крекинга и деалкилирования (распад по С–С-связи) высокомолекулярных молекул исходного сырья с образованием низкомолекулярных (н. м.) углеводородов: а) крекинг парафинов с образованием н. м. парафина и олефина: СnH2n+2 → CmH2m + CpH2p+2 ; б) крекинг олефинов с образованием н. м. олефинов: СnH2n → CmH2m + CpH2p ; в) деалкилирование алкилароматических углеводородов: ArCnH2n+1 → ArH + CnH2n → ArCmH2m–1 + CpH2p ; г) крекинг нафтенов с образованием олефинов: ц-СпН2n → CmH2m + CpH2p , где n = m + р. Первичные реакции распада могут осуществляться либо термически по радикально-цепному механизму, либо каталитически на апротонных (льюисовских) центрах алюмосиликатной матрицы ЦСК: RH + L → R+ + LH– R+ → н. м. олефин + R+' R+' + LH → R'H + L или R+' → H+ + олефин 2. Вторичные бимолекулярные реакции углеводородов на поверхности цеолита с участием карбений-ионов, образующихся преимущественно присоединением протона к олефину (инициирование цепи): RCH = CH2 + HA → RCHCH3 + A— Различие по реакционной способности образующихся карбкатионов обусловливает вероятные направления превращений и степень участия их в дальнейших реакциях. Установлено, что стабильность карбениевых ионов возрастает в ряду: СН3 < +С2Н5 < + первичный < вторичный < третичный. Третичный карбениевый ион является самым стабильным. Именно этим обусловлен высокий выход изопарафиновых углеводородов, особенно изобутана, при каталитическом крекинге. Реакции развития цепи включают следующие наиболее характерные реакции карбениевых ионов: распад С–С-связи, перенос гидридиона (Н-перенос), изомеризацию, циклизацию, дециклизацию, деалкилирование, алкилирование, полимеризацию, поликонденсацию и др. Обрыв цепи превращений карбениевых ионов происходит возвратом протона к поверхности катализатора или отнятием электрона от центров Льюиса. Распад С–С-связи карбений-иона является одной из наиболее важных целевых реакций, приводящих к образованию низкомолекулярных крекинга. Для этой реакции применимы следующие правила: а) легче всего разрывается С–С-связь, находящаяся в β-положении по отношению к атому углерода, несущему заряд (правило — β-распада); б) у образующихся олефинов имеется двойная связь у первого углеродного атома; в) из нескольких возможных вариантов более вероятен β-распад карбений-иона с образованием олефина с меньшей длиной цепи: 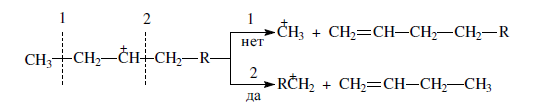 Продукт первичного β -распада — карбений-ион может снова крекироваться до образования более стабильных карбкатионов или углеводородов (после отдачи протона или присоединения электрона); г) более выгодным для алкилароматических или алкилнафтеновых углеводородов является отрыв всей алкильной группы: 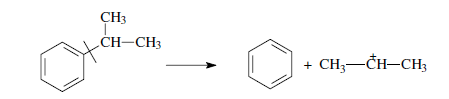 Поскольку образование требует высоких энергетических затрат, цепной распад карбкатионов прерывается до образования карбениевых ионов с числом углеродных атомов 3…5. Перенос гидрид-иона (Н-перенос) можно проиллюстрировать следующим образом: Установлено, что лучшими гидридными донорами являются нафтены, полициклические нафтены или нафтено-ароматические углеводороды, изоалканы и даже олефины. Энергетически более выгоден отрыв гидрид-иона от третичного, затем вторичного и менее выгоден от первичного углеродного атома. Нафтеновые, алкилароматические и изопарафиновые углеводороды часто содержат третичные атомы углерода и поэтому интенсивно участвуют в реакциях Н-переноса. Активными акцепторами гидрид-ионов являются наименее стабильные высокореакционноспособные карбений-ионы или углеводороды, содержащие несколько π-связей, например диолефины. Именно Н-перенос обусловливает повышенные выход топливных фракций и химическую стабильность бензинов каталитического крекинга. По Н-переносу осуществляются следующие реакции каталитического крекинга: олефин + нафтен парафин + арен олефин + парафин парафин + олефин олефин + олефин арен + парафин олефин + олефин арен + водород арен + арен кокс + парафин + водород и т. д. Изомеризация карбениевых ионов является наряду с распадом важной целевой реакцией, повышающей товарные качества продуктов каталитического крекинга. В большинстве случаев изомеризация протекает быстрее, чем крекинг, и потому часто предшествует β-распаду. Сочетание реакций изомеризации и β-распада обусловливает повышенное содержание в продуктах каталитического крекинга углеводородов изостроения. Изомеризация карбениевых ионов может происходить либо путем передачи протона (гидридный сдвиг), либо метильной группы (скелетная изомеризация) вдоль углеводородной цепи: 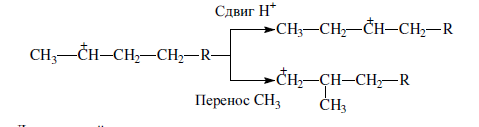 Для реакций изомеризации предложен механизм, согласно которому процесс осуществляется через образование промежуточных циклических структур, например циклопропана, циклобутана и т. д. (по-видимому, посредством многоточечной, т. е. мультиплетной хемосорбции): 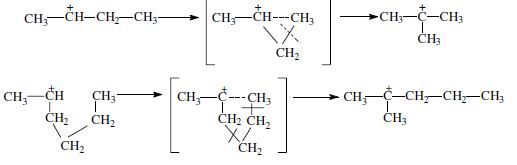 и переносом метильной группы внутри молекулы при изомеризации ди- и полиметилбензолов. Так, ксилолы подвергаются взаимопревращению: 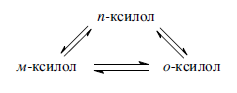 Циклизация и дециклизация как обратимые реакции с участием карбений-ионов протекают, по-видимому, через мультиплетную хемосорбцию: 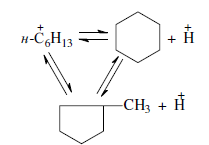 или через диеновый синтез: 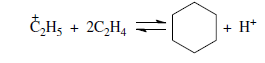 Циклопентаны в условиях каталитического крекинга более устойчивы, чем циклогексаны. Циклогексаны в этих условиях могут подвергаться дегидрированию в арены посредством Н-переноса. При наличии длинных боковых цепей в циклоалкановом карбениевом ионе возможны изомеризация боковой цепи и деалкилирование. Бициклические циклоалкановые карбениевые ионы ароматизируются в большей степени, чем моноциклические. Алкилирование и полимеризация — реакции, противоположные крекингу, протекают по карбений-ионному механизму. При температурах ниже 400 °С они доминируют над крекингом, а при высоких температурах равновесие смещается в сторону деалкилирования и деполимеризации. Конденсация ароматических углеводородов, дающая соединения с более высокой молекулярной массой, вплоть до кокса, характерна для каталитического крекинга. При этом ареновый карбений-ион вступает в последовательные реакции присоединения (конденсации) к ароматическим углеводородам и Н-переноса. Процесс конденсации вследствие высокой стабильности полициклического аренового карбений-иона может продолжаться до обрыва цепи: 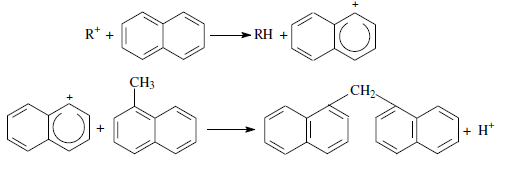 Коксообразование. При осуществлении реакций углеводородов на кислотных катализаторах образуется углеродистый материал, называемый коксом, который не десорбируется с поверхности катализатора. Этот материал имеет атомное отношение водорода к углероду от 0,3 до 1,0 и спектроскопические характеристики, аналогичные таковым для полициклических ароматических соединений. При крекинге ароматических углеводородов кокс получается более обогащенным углеродом, чем при крекинге парафинистого сырья. В составе кокса крекинга сернистого нефтяного сырья всегда содержится сера. В среднем отношение содержания серы в коксе к ее содержанию в сырье крекинга близко к единице. Вследствие экранизации активных центров ЦСК коксовыми отложениями активность катализатора крекинга быстро снижается. Эта дезактивация является обратимой, так как после окислительной регенерации первоначальная активность практически полностью восстанавливается. 2.1.3 Промышленный марки катализаторов каталитического крекинга На отечественных установках с движущимся слоем шарикового катализатора применялись и продолжают пока применяться шариковые катализаторы АШНЦ-3 (без РЗЭ), АШНЦ-6, Цеокар-2 и Цеокар-4 (все с РЗЭ). Из микросферических ЦСК применение находят: КМЦР-2 (2 % La2O3), МЦ-5 и РСГ-6Ц (по 4 % La2O3), КМЦР-4 (с промотором дожига) и др. Из зарубежных ЦСК более известны следующие марки катализаторов: Дюрабед (5, 6, 8, 9), Супер (Д, экстра Д), (1–7), CBZ (1–4),Октакэт-11, Резидкэт (20, 30) и другие. 2.2 Катализаторы каталитического риформинга 2.2.1 Состав катализаторов каталитического риформинга и механизм Процесс каталитического риформинга осуществляют на бифункциональных катализаторах, сочетающих кислотную и гидрирующую-дегидрирующую функции. Гомолитические реакции гидрирования и дегидрирования протекают на металлических центрах платины или платины, промотированной добавками рения, иридия, олова, галлия, германия и др., тонко диспергированных на носителе. Кислотную функцию в промышленных катализаторах риформинга выполняет носитель, в качестве которого используют оксид алюминия. Для усиления и регулирования кислотной функции носителя в состав катализатора вводят галоген: фтор или хлор. В настоящее время применяют только хлорсодержащие катализаторы. Содержание хлора составляет от 0,4-0,5 до 2,0 % мас. Бифункциональный механизм доказан на примере использования катализаторов, содержащих только кислотные центры или только металлические центры, которые оказались исключительно малоактивными, в то время как даже механическая их смесь была достаточно активна. Благодаря бифункциональному катализу удается коренным образом преобразовать углеводородный состав исходного бензина и повысить его октановую характеристику на 40...50 пунктов. Платина на катализаторе риформинга не только ускоряет реакции гидрирования-дегидрирования, но и замедляет образование кокса на его поверхности. Обусловливается это тем, что адсорбированный на платине водород сначала диссоциируется, затем активный (атомарный) водород диффундирует на поверхности катализатора к кислотным центрам, ответственным за образование коксовых отложений. Коксогены гидрируются и десорбируются с поверхности. В этой связи скорость образования кокса при прочих равных условиях симбатно зависит от давления водорода. Поэтому минимальная концентрация платины в катализаторах риформинга определяется необходимостью прежде всего поддерживать их поверхность в «чистом» виде, а не только с целью образования достаточного числа активных металлических центров на поверхности носителя. В монометаллических алюмоплатиновых катализаторах риформинга содержание платины составляет 0,3…0,8 % мас. Очень важно, чтобы платина была достаточно хорошо диспергирована на поверхности носителя. С увеличением дисперсности платины повышается активность катализатора. Прогресс каталитического риформинга в последние годы был связан с разработкой и применением сначала биметаллических и затем полиметаллических катализаторов, обладающих повышенной активностью, селективностью и стабильностью. Используемые для промотирования металлы можно разделить на две группы. К первой из них принадлежат металлы 8-го ряда: рений и иридий, известные как катализаторы гидродегидрогенизации и гидрогенолиза. К другой группе модификаторов относят металлы, практически неактивные в реакциях риформинга, такие как германий, олово и свинец (IV группа), галлий, индий и редкоземельные элементы (III группа) и кадмий (из II группы). К биметаллическим катализаторам относят платино-рениевые и платино-иридиевые, содержащие 0,3…0,4 % мас. платины и примерно столько же Re и Ir. Рений или иридий образуют с платиной биметаллический сплав, точнее кластер, типа Pt-Re-Re-Pt-, который препятствует рекристаллизации — укрупнению кристаллов платины при длительной эксплуатации процесса. Биметаллические кластерные катализаторы (получаемые обычно нанесением металлов, обладающих каталитической активностью, особенно благородных, на носитель с высокоразвитой поверхностью) характеризуются, кроме высокой термостойкости, еще одним важным достоинством — повышенной активностью по отношению к диссоциации молекулярного водорода и миграции атомарного водорода (спилловеру). В результате отложение кокса происходит на более удаленных от металлических центров катализатора, что способствует сохранению активности при высокой его закоксованности (до 20 % маc. кокса на катализаторе). Из биметаллических катализаторов платино-иридиевый превосходит по стабильности и активности в реакциях дегидроциклизации парафинов не только монометаллический, но и платино-рениевый катализатор. Применение биметаллических катализаторов позволило снизить давление риформинга (от 3,5 до 2...1,5 МПа) и увеличить выход бензина с октановым числом по исследовательскому методу до 95 пунктов примерно на 6 %. Полиметаллические кластерные катализаторы обладают стабильностью биметаллических, но характеризуются повышенной активностью, лучшей селективностью и обеспечивают более высокий выход риформата. Срок их службы составляет 6-7 лет. Эти достоинства их обусловливаются, по-видимому, тем, что модификаторы образуют с платиной (и промоторами) поверхностные тонкодиспергированные кластеры с кристаллическими структурами, геометрически более соответствующими и энергетически более выгодными для протекания реакций ароматизации через мультиплетную хемосорбцию. Среди других преимуществ полиметаллических катализаторов следует отметить возможность работы при пониженном содержании платины и хорошую регенерируемость. Успешная эксплуатация полиметаллических катализаторов возможна лишь при выполнении определенных условий: — содержание серы в сырье риформинга не должно превышать 1 · 10–4 % маc., для чего требуется глубокое гидрооблагораживание сырья в блоке предварительной гидроочистки; — содержание влаги в циркулирующем газе не должно превышать (2-3) · 10–3 % мольн.; — при пуске установки на свежем и отрегенерированном катализаторе требуется использование в качестве инертного газа чистого азота (полученного, например, ректификацией жидкого воздуха); — для восстановления катализатора предпочтительно использование электролитического водорода. 2.2.2 Механизм и химизм процесса каталитического риформинга Целевыми в процессах каталитического риформинга являются реакции образования ароматических углеводородов за счет: 1) дегидрирования шестичленных циклоалканов 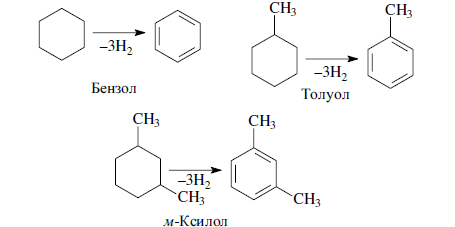 2) дегидроизомеризации циклопентанов 3) дегидроциклизации (С5- или С6-дегидроциклизации) парафиновых углеводородов 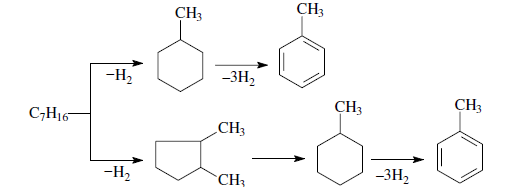 Схему реакций дегидроциклизации н-гептана можно представить и в следующем виде: 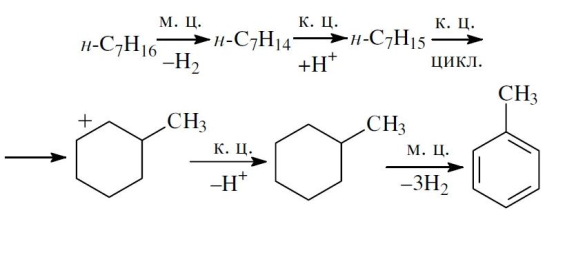 2.2.3 Марки катализаторов каталитического риформинга В настоящее время отечественной промышленностью вырабатываются три типа катализаторов риформинга : – монометаллические (АП-56 и АП-64); – биметаллические (КР-101 и КР-102); – полиметаллические (КР-104, КР-106, КР-108 и платино-эрионитовый СГ-ЗП). Список использованной литературы Ахметов С.А. Технология глубокой переработки нефти и газа: Учебное пособие для вузов. Уфа: Гилем, 2002. 672 с. Магарил Р.З. Теоретические основы химических процессов переработки нефти: Учебное пособие для вузов.- Л.: Химия. 1985, 280 с. |