Лабораторная работа номер 1. Отчет по лабораторной работе 1 по дисциплине Химия нефти и газа
 Скачать 441.5 Kb. Скачать 441.5 Kb.
|

Инженерная школа природных ресурсов Направление 21.03.01 Нефтегазовое дело Отделение нефтегазового дела Специализация – «Эксплуатация и обслуживание объектов транспорта и хранения нефти, газа и продуктов переработки» ОТЧЕТ по лабораторной работе № 1 по дисциплине: Химия нефти и газа Студент, группы 2Б11 В. В. Кондратьев (подпись, дата) Преподаватель Ассистент ОНД ИШПР С.Н. Джалилова _______________ оценка Томск – 2022 ОПРЕДЕЛЕНИЕ КОМПОНЕНТНОГО СОСТАВА ГАЗОВ МЕТОДОМ ХРОМАТОГРАФИИ В настоящее время лидирующее положение при исследовании состава нефти, конденсатов, нефтепродуктов, природных и попутных газов, сжиженного газа получили физико-химические методы анализа, в том числе, хроматография. Физические основы и аппаратная реализация метода газовой хроматографии Газовая хроматография как эффективный метод разделения и анализа сложных смесей газов, жидкостей и твердых тел получила широкое признание в 50-х годах нашего столетия и с тех пор непрерывно развивается и совершенствуется. Термин «хроматография» происходит от греческих слов chromatos -цвет, окраска и grapho- пишу, описываю. В 1903-1906 гг. русский ученый-ботаник М.С. Цвет в результате экспериментов разделил сложную смесь растительных пигментов из листьев растений при пропускании ее петролейно-эфирного раствора через вертикальную стеклянную колонку, заполненную порошкообразным карбонатом кальция. При этом возник ряд окрашенных зон, по числу которых можно было судить о сложности состава анализируемой смеси. Пропуская через колонку различные растворители (полярные, неполярные), оказалось возможным регулировать степень распределения зон по длине колонки: сдвигать или раздвигать их, тем самым способствуя повышению точности последующего качественного и количественного определения. Так была создана жидкостная адсорбционная хроматография. В дальнейшем в качестве подвижной фазы стали использовать не только жидкость, но также пар или газ. Любую разновидность хроматографии можно определить как динамический метод разделения смеси веществ, основанный на многократно повторяющемся процессе перераспределения компонентов между двумя несмешивающимися фазами, одна из которых является неподвижной, а другая - подвижной: неподвижная фаза - твердый адсорбент или суспензия адсорбента в жидкости, или жидкость, наносимая на поверхность твердого носителя; подвижная фаза - газ или жидкость, протекающие вдоль слоя неподвижной фазы. Понятие (термин) газовая хроматография объединяет все методические варианты хроматографии, в которых подвижная фаза газообразна. К газожидкостной (распределительной) хроматографии (ГЖХ) относятся все методические варианты газовой хроматографии, в которых в качестве неподвижной фазы используется слой жидкости, нанесенный на поверхность твердого носителя (зернистый мелкодисперсный материал или внутренние стенки колонки). Газоадсорбционная хроматография (ГАХ) включает все методические варианты газовой хроматографии, в которых неподвижной фазой является активное дисперсное твердое тело (адсорбент); древесный уголь, силикагсль, графитированная сажа и др. Имеется возможность использовать одновременно оба способа в одной колонке, когда в качестве наполнителя применяется так называемый модифицированный адсорбент, представляющий собой твердый адсорбент сравнительно небольшой активности, на который нанесена какая-либо жидкость (например, вазелиновое масло) в количестве, недостаточном дня заполнения всей поверхности адсорбента. Жидкость расположится на наиболее активных ее центрах. Такой модифицированный адсорбент обладает в определенной степени свойствами и твердого адсорбента и нанесенной жидкости. В связи с тем, что многочисленные варианты хроматографии в настоящее время используются и для решения неаналитических задач, хроматографию в целом можно определить и как «область науки, изучающую процессы, основанные на перемещении зоны вещества вдоль слоя сорбента в потоке подвижной фазы и связанные с многократным повторением сорбционных и десорбционных актов». Принципиальная схема газохроматографического анализа следующая. Перед началом анализа хроматографическую колонку, содержащую неподвижную фазу, непрерывно промывают практически несорбирующимся (инертным) газом. Затем в этот газ-носитель у входа в колонку вводят порцию (дозу) анализируемой смеси компонентов, например, А, В иС. Вследствие различий в сорбции или растворимости при движении через слой неподвижной фазы компоненты группируются в зоны, отделенные друг от друга инертным газом носителем G(рис. 1, а). Из-за диффузионных процессов в подвижной и неподвижной фазах границы зон размываются, поэтому максимальная концентрация каждого компонента оказывается сосредоточенной в центре зоны. Если на выходе из колонки регистрировать изменение во времени какого-либо физического свойства газового потока (так называемое дифференциальное детектирование), то выходная хроматографическая кривая - хроматограмма - запишется в виде пиков, возвышающихся над нулевой (базовой) линией (рис. 1, 6).  Рис.1. Проявительная газовая хроматография: а - участок колонки с распределением хроматографических зон; б – хроматограмма Времена выхода компонентов, отсчитываемые от момента ввода пробы до момента регистрации вершины пика, или объемы газа-носителя, затраченные на перенос через колонку каждого компонента, дают качественную характеристику анализируемых веществ. Сопоставление площадей (или высот) хроматографических пиков позволяет с высокой точностью выполнять количественные определения. Одним из недостатков газохроматографического анализа при постоянных температуре и скорости газа-носителя является то, что, анализируя смесь компонентов, сильно различающихся по характеристикам удерживания, трудно выбрать оптимальные температуру колонки и скорость газа-носителя. При невысокой температуре колонки (или небольшой скорости газа-носителя) лишь пики первых, как правило, наиболее летучих компонентов, будут резко очерчены на хроматограмме. Пики последующих компонентов, вследствие все большего размывания потоком газа-носителя, будут регистрироваться на хроматограмме все более широкими и в пределе могут слиться с нулевой линией. Общая продолжительность анализа при этом составит довольно значительное время. При повышенной температуре колонки или увеличенной скорости газа-носителя четко пропишутся на хроматограмме пики наименее летучих соединений пробы, последними выходящие из колонки. Общее время анализа будет небольшим, однако наиболее летучие (наименее удерживаемые) компоненты выйдут из колонки частично или полностью неразделенными. Принципиальная схема хроматограф Газовый аналитический хроматограф представляет собой совокупность взаимодействующих систем, предназначенных для проведения анализа в оптимальном режиме хроматографического разделения исследуемой смеси с целью определения ее состава. Газовый хроматограф состоит из следующих основных частей: системы подготовки газа-носителя, дозатора, хроматографической колонки, детектора, системы термостатирования. Принципиальная (функциональная) схема аналитического лабораторного газового хроматографа представлена на рис. 2. Газ-носитель из баллона высокого давления 1 через регулятор расхода 2, захватив из крана-дозатора или испарителя пробу анализируемой смеси, направляется в хроматографическую колонку 5. После колонки газ-носитель вместе с компонентом смеси поступает в детектор 6 и далее - в атмосферу. Детектор преобразует изменение физических или физико-химических свойств бинарных смесей (компонент - газ-носитель по сравнению с чистым газом-носителем) в электрический сигнал, который регистрируется самописцем 7. Температура колонки и детектора поддерживается постоянной термостатами 4. Современный хроматограф серии «Кристалл» состоит из аналитического блока и станции управления, контроля и обработки хроматографической информации, в качестве которой используется компьютер и специальная программа (рис. 3). Один компьютер может работать в реальном времени с несколькими аналитическими блоками. 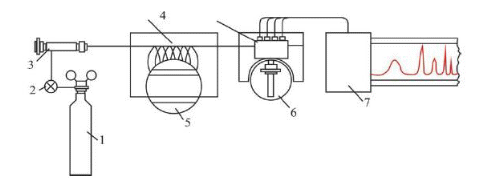 Рис. 2. Принципиальная схема газового хроматографа: 1-баллон с газом-носителем; 2-регулятор расхода; 3-место ввода пробы (кран-дозатор, испаритель); 4- термостаты; 5-колонка; 6-детектор; 7- регистратор Хроматограф полностью автоматизирован, начиная от ввода пробы и заканчивая обработкой хроматографической информации, в т. ч. реализованы функции автоматического регулирования температуры термостатов, расходов газа-носителя и вспомогатетьных газов, автоматического поджига детекторов и контроля горения пламени в процессе работы; измерения сигналов детекторов.  Рис. 3. Хромапюграфический комтекс серии «Кристаллюкс» Система подготовки газа-носителя Газ-носитель из баллона загрязнен примесями кислорода, воды и органических соединений. Система детектирования, природа разделяемых веществ и неподвижной жидкой фазы предъявляют жесткие требования к чистоте газа-носителя. Незначительные примеси кислорода приводят к изменению характеристик сорбента и изменяют времена удерживания. Поэтому требуется тщательная очистка газа от примесей. Кислород удаляется с помощью катализаторов при комнатной температуре. Молекулярные сита хорошо очищают газ от паров воды. Органические соединения удаляются активированным углем. На характеристики удерживания компонента сильное влияние оказывает скорость газа-носителя. Чтобы исключить колебания скорости потока во время опыта, хроматографы снабжаются устройствами для стабилизации и измерения скорости газа-носителя. Расход газа устанавливают с помощью дросселей. Стабилизация расхода газа осуществляется регулятором давления или регулятором расхода. Точное значение скорости газового потока, проходящего через колонку, необходимо знать для вычисления параметров удерживания. Расход газа-носителя на выходе из колонки периодически измеряют пенным расходомером. Для непрерывного наблюдения за скоростью газового потока предусмотрены ротаметры. Дозирующие устройства На эффективность разделения влияет величина и способ ввода пробы в хроматограф. При введении пробы необходимо обеспечить идентичность ее состава с анализируемой смесью. Нарушение идентичности может быть вызвано потерей части пробы при введении ее в колонку (например, вследствие испарения), наличием в дозаторе непродуваемых («мертвых») объемов и другими причинами. Величина пробы выбирается с учетом чувствительности детектора и сорбционной емкости колонки. Объем или масса пробы должны воспроизводиться в пределах 1-3 %. Проба должна вводиться в колонку по возможности мгновенно, чтобы уменьшить размывание пиков на хроматограмме и не нарушить установившийся режим хроматографа. Для дозирования и ввода газообразных смесей применяют краны-дозаторы. Объем сменных калиброванных петель позволяет вводить пробы от 0,1 до 10 мл. Жидкие пробы вводят в колонку с помощью специальных микрошприцев через термостойкое резиновое уплотнение испарителя. Объем пробы в зависимости от типа детектора колеблется в пределах 0,1-50 мкл. Хроматографические колонки Различают три основных типа аналитических колонок - насадочные (набивные), микронасадочные и капиллярные (рис. 4). Эффективность работы насадочных колонок зависит от типа и количества жидкой фазы, размера частиц твердого носителя и метода заполнения колонки. Капиллярные колонки для ГЖХ представляют собой трубки диаметром 0.2-0,5 мм, внутренние стенки которых покрыты тонким слоем жидкой фазы. Длина таких колонок от 10 до 100-200 м. Эффективность капиллярных колонок доходит до 1000 теоретических тарелок на метр длины. 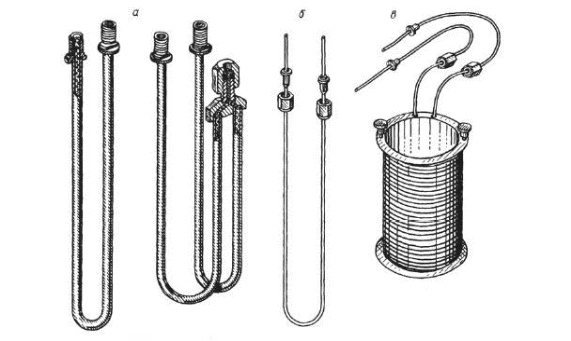 Рис. 4. – Хроматографические колонки: а-насадочная колонка, б-микронасадочная, в-капилярная колонка. Детекторы Хроматографический детектор - это устройство, предназначенное для обнаружения и количественного определения выходящих из колонки в потоке газа-носителя компонентов анализируемой смеси. В газовой хроматографии чаше используют дифференциальные детекторы, которые в отличие от интегральных измеряют мгновенную концентрацию компонента в потоке газа-носителя. В настоящее время создано несколько десятков типов детекторов. В современных хроматографах применяют детекторы, использующие некоторые физические свойства газа, такие как: теплопроводность, плотность, теплота сгорания, способность молекул газа ионизироваться (приобретать электрический заряд) и некоторые другие. Во всех случаях используется различие физических свойств газа-носителя, с одной стороны, и компонентов газа, с другой. Если через детектор проходит только газ-носитель, то детектор не реагирует (его сигнал равен нулю); как только в него начнет поступать газ-носитель с каким-либо компонентом анализируемой смеси, то возникает сигнал, пропорциональный концентрации компонента в газе-носителе. Подчеркнем, что поскольку детектор установлен после хроматографической колонки, то он имеет дело уже не со сложной многокомпонентной смесью, а лишь с чистым газом-носителем или его смесью с одним из компонентов пробы газа. Один из наиболее широко используемых детекторов - детектор по теплопроводности или катарометр. В нем для обнаружения в потоке газа-носителя компонентов пробы используется различие теплопроводности газа-носителя и компонента. Типовая конструкция представляет собой массивный металлический корпус 6 (рис. 5), в котором имеются две камеры: сравнительная 1 и измерительная 2. В них находятся проволочные или полупроводниковые сопротивления R2обладающие относительно большими температурными коэффициентами и представляющие собой два плеча схемы моста Уитстона. Камеры детектора включены в газовую схему хроматографа следующим образом: Газ-носитель с постоянной скоростью поступает в сравнительную камеру 1 детектора, откуда проходит через канат крана 3 или через пробоотборный объем 5 в хроматографическую колонку 4 и далее через измерительную камеру 2 детектора выходит наружу. Схема моста питается постоянным током, но в отличие от традиционных мостовых измерений ток питания схемы велик, в результате чего сопротивления R 3 и R2нагреваются и их температура будет выше, чем у окружающих их металлических стенок камер. Часть тепла нагретых сопротивлений передается окружающим стенкам, главным образом, благодаря теплопроводности газа-носителя. При постоянных условиях нагрева сопротивлений (постоянная величина тока питания детектора), постоянном расходе газа-носителя (поддерживаемым регулятором) и постоянной температуре корпуса детектора (для чего он термостатируется) через некоторое время в обеих камерах установится тепловое равновесие, при котором сопротивления R3и R2будут иметь постоянную температуру, превышающую температуру стенок детектора обычно на 30-50 °С. Величина этих сопротивлений будет также постоянной, и установится равновесие измерительной схемы моста Уитстона.  Рис. 5.- Принципиальная схема хроматографа с детектором по теплопроводности Такое равновесие, фиксируемое регистратором в виде «нулевой линии», будет до тех пор, пока все перечисленные факторы будут неизменными, т.е. пока через обе камеры проходит только газ-носитель с определенной, свойственной данному газу, теплопроводностью. В тот момент, когда из колонки выделится первый компонент пробы (например, метан), имеющий другую (пусть в данном случае меньшую) величину теплопроводности, чем газ-носитель, то и смесь его с газом-носителем будет иметь меньшую теплопроводность. Когда смесь попадает в измерительную камеру детектора, в ней нарушается тепловой режим, поскольку меньшее количество тепла будет переноситься новой газовой смесью на стенки камеры детектора. В результате температура данного плеча моста R2 повысится, а, следовательно, увеличится его сопротивление, и равновесие схемы нарушится (мост разбалансируется), что и зафиксирует регистратор, как отклонение его указателя. Через некоторое время из колонки вновь будет выходить только газ-носитель, восстанавливая первоначальные условия в измерительной камере. Указатель регистратора постепенно вернется в прежнее (нулевое) положение и таким образом на ленте прибора будет записана кривая в виде пика. Выход из колонки следующего компонента пробы (например, этана) создаст такой же характер изменений в измерительной камере и будет записан новый пик (этана) и т.д. до тех пор, пока из колонки не выйдут все составляющие исследуемую пробу компоненты. В результате будет записана кривая в виде ряда пиков, называемая хроматограммой (рис. 6). 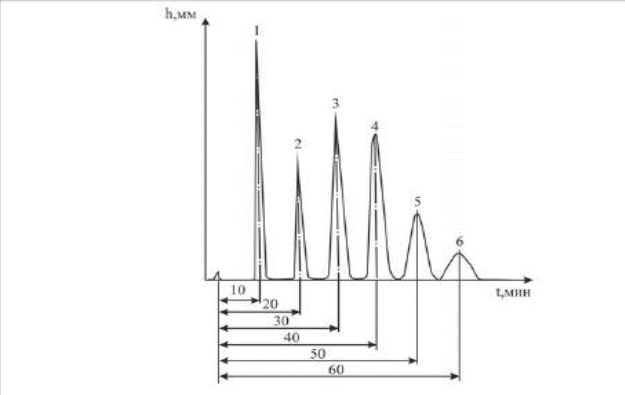 Рис. 6 – Хроматограмма. Идентификация компонентов. Величина сигнала зависит как от природы компонента, так и от содержания его в анализируемой смеси. Чем выше концентрация компонента, тем больше будет пик, поскольку резче изменятся условия в измерительной камере. Находят применение и другие виды детекторов: Детектор по теплоте сгорания (термохимический), использует эффект теплоты сгорания компонентов анализируемой пробы в присутствии катализатора - платинового проволочного сопротивления, являющегося одновременно и чувствительным элементом детектора. Детектор по плотности газов (денситометрический). В этом детекторе используется различие плотностей газа-носителя и компонентов анализируемой пробы. Пламенно-ионизационный детектор. Ионизация анализируемых веществ происходит в процессе их сгорания в пламени водорода, что вызывает соответствующее возрастание ионного тока. Система термостатирования Хроматографические колонки, детекторы, испарители работают при определенных температурных режимах. Выбранная температура колонки должна поддерживаться постоянной с погрешностью, не превышающей 0.2 °С. Точность поддержания температуры детектора зависит от его типа. Для катарометра требуется более стабильное термостатирование, чем для колонки: максимальные колебания температуры не более 0,02 °С. Пламенно-ионизационный детектор может устойчиво работать без специального термостата. Требуемые температурные режимы колонки, детектора и дозирующих устройств достигаются помещением их в соответствующие термостаты, управляемые терморегулятором. Если необходимо повышать температуру колонки в процессе анализа, используют программатор температуры. Хроматографы снабжаются воздушными термостатами с вентиляторами. Термостаты, предназначенные для работы с программированием температуры, имеют меньшую теплоемкость, так как должны быстро прогреваться при ограниченной мощности нагревателей. Программирование температуры Программирование температуры колонки применяется при анализе сложных смесей с широким диапазоном температур кипения (более 100 К). Общее время анализа значительно сокращается по сравнению с работой в изотермическом режиме. Чаще всего используется линейный закон (постоянная скорость повышения температуры) или линейно-ступенчатый режим, при котором участки повышения температуры чередуются с изотермическими ступенями. Система программирования обеспечивает скорости нагрева от 0,5 до 25 °С/мин. Линейное программирование осуществляется с помощью электродвигателя, изменяющего сопротивление потенциометра задания температуры и одновременно поворачивающего температурную шкалу программатора. Необходимая скорость нагрева достигается установкой соответствующего питания двигателя программатора. Термостаты и терморегулятор с программатором составляют систему термостатирования, в которую может также входить устройство для измерения температуры. Регистрация результатов анализа Сигнал детектора, преобразованный усилителем, записывается в виде хроматограммы автоматическим регистратором: дисплеем или потенциометром. Обычно регистрируется зависимость величины сигнала детектора от времени. Хроматограмма природного горючего газа, полученная на хроматографе серии «Кристалл», приведена на рис. 7. 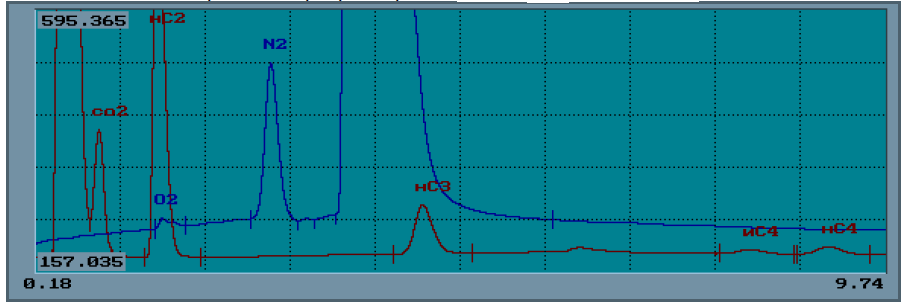 Рис. 7 – Хроматограмма природного горючего газа На хроматограмме одному компоненту всегда будет соответствовать один пик. Однако нельзя сказать, что одному пику всегда соответствует только один компонент. Если из колонки одновременно выделились два компонента (не разделившись), то они также зарегистрируются в виде одного пика. Газ-носитель Природа газа-носителя оказывает влияние на работу детектора и характеристики колонки. Кроме обеспечения высокой чувствительности детектора, газ-носитель должен быть инертным по отношению к разделяемым веществам и сорбенту, иметь небольшую вязкость для поддержания минимального перепада давления на колонке, быть взрывобезопасным и достаточно дешевым. Детектор по теплопроводности измеряет различие в теплоопроводности чистого газа-носителя и смеси газа-носителя с веществом, выходящим из хроматографической колонки. Поэтому наибольшая чувствительность может быть получена в том случае, когда теплопроводность анализируемого вещества сильнее отличается от теплопроводности газа-носителя. Большинство органических веществ имеют низкую теплопроводность и для их анализа целесообразно использовать газы-носители с возможно более высокой теплопроводностью. Такими газами являются водород и гелий, но на практике водород ввиду его взрывоопасности применяется значительно реже гелия. Так как гелий является довольно дефицитным и дорогим газом, а работа с водородом небезопасна, в некоторых случаях в качестве газов-носителей могут использоваться азот, аргон, углекислый газ или воздух. Однако характеристики детектора по теплопроводности (чувствительность, линейность) при работе с этими газами значительно ухудшаются. Кроме того, при анализе веществ с большей теплопроводностью, чем у газа-носителя, появляются отрицательные пики. Теплопроводность газов зависит от подвижности их молекул. Скорость молекул является функцией молекулярного веса: чем меньше молекула, тем больше ее скорость и тем выше теплопроводность газа. Поэтому водород и гелий, имеющие наименьшие размеры молекул, обладают самой большой теплопроводностью. Неподвижная фаза Эффективность хроматографического разделения во многом зависит от выбора неподвижной фазы. Правильный выбор неподвижной фазы, в свою очередь, зависит от природы анализируемого вещества. При выборе стационарной фазы следует учитывать, что неполярные вещества обычно лучше разделяются на неполярных фазах. Сильное влияние на качество разделения оказывают водородные связи, которые возникают между анализируемым веществом и жидкой фазой. На процесс разделения влияют и донорно-акцепторные связи. Таким образом, основным фактором, определяющим качество разделения, является правильный выбор неподвижной фазы. В газоадсорбционной хроматографии в качестве стационарной фазы широко применяются пористые полимеры (в виде шариков) – полисорбы, молекулярные сита, активированный уголь, оксид алюминия, силикагель. К неполярным сорбентам относят активированный уголь, различные сорта которого отличаются размерами пор. Силикагель, оксид алюминия, молекулярные сита применяют как полярные адсорбенты. Сополимеры стирола и дивинилбензола (полисорбы) подразделяют на сорбенты средней полярности и неполярные (за рубежом аналогичные адсорбенты выпускаются под названием порапаки). В газожидкостной хроматографии неподвижной фазой служит жидкости, нанесенная на твердый носитель, который должен отвечать определенным требованиям: быть химически инертным, термостойким, механически прочным и не обладать адсорбционной активностью. Число рекомендуемых жидких фаз в настоящее время очень велико: несколько сот. В целях систематизации жидкие фазы поделены на 14 классов. К важнейшим неподвижным фазам относятся: сквалан, апиезоны. силиконовые масла, силиконовые смазки, производные углеводородов и др. Оптимальная жидкая фаза, как правило, подбирается опытным путем. Наличие большого количества наполнителей колонок делает хроматографический метод анализа газов в значительной степени универсальным. В настоящее время имеется возможность проводить по этому методу анализы очень большого количества веществ (сильно отличающихся по своим физическим свойствам), начиная с легких газов (например, водород, кислород, азот), до тяжелых углеводородов с количеством углеродных атомов выше 30. При работе с жидкими (при стандартных температуре и давлении) углеводородами последние перед тем, как попасть в хроматографическую колонку, должны быть предварительно испарены. Следовательно, в этом случае уже будем иметь дело с парами веществ. Дальнейшее разделение их будет происходить так же как и разделение газов. Адсорбенты, применявшиеся первоначально как единственные наполнители (пока не была предложена газожидкостная хроматография), остались и теперь основными, для анализа легких газов и углеводородов, включая фракцию С2. Более тяжелые углеводородные газы лучше разделяются способом газожидкостной хроматографии, благодаря его указанным выше преимуществам. Последние не могут быть использованы для легких газов потому, что не удается подобрать такие жидкости, в которых они имели бы необходимую для целей разделения растворимость, оставаясь при этом достаточно стойкими и с малой упругостью пара. С другой стороны, жидкости, так хорошо используемые для углеводородных фракций С3-С10, имеют принципиальный недостаток: при значительном повышении температуры колонки, что необходимо для разделения углеводородов с высокой температурой кипения, их упругость пара становится значительной и, следовательно, они, испаряясь, быстро выносятся из колонки газом-носителем. В связи, с расширением пределов анализируемых веществ в сторону возрастания их температур кипения, становится все труднее подобрать нужную жидкую фазу для разделения. Среда применяемых в настоящее время адсорбентов особое значение приобрели так называемые молекулярные сита. Они представляют собой искусственные цеолиты с различной структурной решеткой и с порами определенного размера, причем того же порядка величины, что и размеры молекул, разделяемых с помощью их компонентов. Эти поры ведут в полости с очень большой поверхностью, но последняя становится доступной лишь тем молекулам газа, размеры которых позволяют им проникнуть внутрь этих полостей через узкие поры. Молекулярные сита изготавливают нескольких типов и применяют в основном для разделения смесей легких газов, таких, как кислород, азот, аргон, водород, гелий, окись углерода и углеводороды до этана включительно. Молекулярные сита благодаря специальной технологии их изготовления обладают большой стабильностью своей структуры, а, следовательно, и своих адсорбционных свойств, при условии, если их не увлажнять и не засорять более тяжелыми углеводородами. ЛАБОРАТОРНАЯ РАБОТА №1 ОПРЕДЕЛЕНИЕ ФРАКЦИОННОГО, УГЛЕВОДОРОДНОГО И ГРУППОВОГО СОСТАВОВ ДИСТИЛЛЯТА ГАЗОВОГО КОНДЕНСАТА МЫЛЬДЖИНСКОГО МЕСТОРОЖДЕНИЯ ГАЗОХРОМАТОГРАФИЧЕСКИМ МЕТОДОМ ЦЕЛЬ РАБОТЫ: Определить фракционный, углеводородный и групповой состав, октановое число дистиллята газового конденсата Мыльджинского месторождения газохроматографическим методом. СУЩНОСТЬ МЕТОДА: Определение состава дистиллята с помощью пламенно-ионизационного детектора (ДИП). Принцип его действия основан на ионизации молекул анализируемых органических соединений в водородном пламени с последующим измерением ионного тока. Сигнал детектора прямо пропорционален количеству анализируемого вещества, поступающего в него в единицу времени. Для работы ДИП необходимы следующие газы: водород, который смешивается с элюатом и сгорает при выходе из горелки, и воздух, обеспечивающий горение водорода. воздух вводится и с помощью диффузора поступает к горелке. Сгорая в воздухе, водород почти не образует ионов, поэтому электропроводность чистого водородного пламени очень низкая (сопротивление пламени 1014Ом) и ток в цепи чрезвычайно мал (10-11– 10-12 А). Этот ток называют фоновым. Как только в водородное пламя попадают органические соединения, они (или продукты их горения) легко ионизируются, в результате чего электропроводность пламени резко возрастает. В цепи двух электродов возникает ионный ток, сила которого зависит от количества молекул органического вещества, поступающих в пламя вместе с водородом в единицу времени. Этот ток очень мал; он увеличивается усилителем и подается на самописец. Для зажигания пламени в горелке есть специальный элемент, находящийся рядом с ней. Чтобы пламя в детекторе не погасло, имеется автоматическая система зажигания пламени, его контроля и сигнализации. Работа пламенно-ионизационного детектора зависит от правильного выбора скоростей газов. Потоки водорода со скоростью 500 мл/мин, воздуха 250 мл/мин и газа-носителя 50 мл/мин обеспечивают равномерное горение с образованием пламени между двумя электродами. Пламенно-ионизационный детектор обладает большой чувствительностью и малой инерционностью. Особенно широко применяется этот детектор в работе с капиллярными колонками и колонками малого диаметра, так как позволяет брать очень малые пробы. Недостатки пламенно-ионизационного детектора: применим только для анализа горючих веществ; не чувствителен к воде, муравьиной кислоте, воздуху, инертным газам, а также к газам и парам CS2, H2S, SO2, NO, NO2, N2O, CO, CO2, SiCl4, SiF4 и др. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ: Включение хроматографа. Ожидание готовности (выход хроматографа на режим): t=35 ºC; VHe=30 мл/мин; Vвозд=200 мл/мин; tиспар=250 ºC; VН2=20 мл/мин. Отбор пробы микрошприцом V= 0,2 мкл. Ввод пробы, старт анализа. В испарителе происходит испарение пробы и проба поступает в колонку. Капиллярная колонка представляет собой неподвижную жидкую фазу, в которой происходит растворение анализируемых углеводородов. Колонка помещена в термостат, где происходит программируемый нагрев (tнач.кол.=35 ºC; tкон.кол.=375 ºC). В результате при определенной температуре происходит испарение углеводородных компонентов в определенном порядке (в соответствии с их свойствами), которые поступают в ДИП, где происходит анализ. Полученные данные регистрируются на ЭВМ с помощью программного обеспечения Хроматек-аналитик 2.5. Далее результаты обсчитываются с помощью внешней программы расчета «Хроматек-Gasoline». ВЫВОДЫ: Получен фракционный состав газохроматографическим методом (таблица 1) Примерно при температуре 55 градусов цельсия начинается более активное выкипание, при дальнейшем нагревании при температуре в 95.30 градусов цельсия — 25 доля отгона. Получили углеводородный состав с помощью ПО «Хроматэк Аналитик 2.5» получено 13 пиков (таблица 2). Из таблицы видно, что основной компонент дистиллята газового конденсата — пропан с концентрацией 52.901. Выделяются из основной массы также пики с компонентами бутан и н-бутан (18.604 и 15.321) После анализа всех пиков был определён групповой состав с помощью программы «Хроматек Газалин 1.1» (таблица 3). Первую тройку по численному значению октанового числа составила 3 группы: нафтены (38922), изопарафины (31457), парафины (22111) соответсвенно. Определено октановое число (таблица 3) по исследовательскому методу октановое число = 77926 куда основной вклад вносят нафтены (31294), парафины (23364) и изопарафины (23364) соответственно. На основные компоненты приходится 7753. |