Валидация фармакопейных методик. Валидация. Вырезки из всего подряд
 Скачать 128.22 Kb. Скачать 128.22 Kb.
|

|
Вырезки из всего подряд Объем валидации МИ • Валидация в полном объеме – если используемые МИ не включены в действующие ТНПА • Валидация в сокращенном объеме – если используемые МИ включены в действующие ТНПА. Объем валидации в данном случае определяется поставленной целью. Все методики и испытания, включенные в фармакопеи стран-участников ICH (ICH - The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use – Международная конференция по гармонизации технических требований к регистрации лекарственных препаратов для использования у человека), являются валидированными. Валидация фармакопейных методик может быть выполнена в меньшем объёме, так как в полном объёме это было выполнено ранее при разработке. Для валидации фармакопейной методики определения количественного содержания (активности) действующих веществ в антибиотиках, в т.ч. микробиологическим методом, обязательными параметрами являются показатели точности: - прецизионность (точность); - правильность, Прецизионность (точность). • Прецизионность МИ - выражение степени близости результатов для серии измерений, выполненных по данной методике на различных пробах одного итого же образца (одной серии). • Прецизионность подтверждается повторяемостью, внутри- и межлабораторной воспроизводимостью. Повторяемость – характеризует точность методики при ее выполнении в течение небольшого промежутка времени в одних и тех же условиях: одним аналитиком, при неизменных условиях окружающей среды (t, W, P). • Используют трехдозный метод диффузии в агар. Тест- микроорганизмы, питательные среды, буферные растворы – в соответствии с ??? • Готовят чашки Петри, как для определения активности антибиотиков методом диффузии в агар трехдозным методом с использованием требуемого тест-микроорганизма • Повторяемость оценивают по результатам 6-ти испытаний по 2 параллельных испытания для каждого из 3-х контейнеров (флаконов) одной серии препарата. Находят: - Стандартное отклонение для каждого определения; - Среднее значение стандартного отклонения (для всех определений) - Относительное стандартное отклонение (RSD) • Критерий приемлемости для подтверждения повторяемости – величина относительного стандартного отклонения (RSD): отношение среднего значения стандартного отклонения к среднему значению активности (количественного содержания) исследуемого вещества для всех определений (%). • RSD – не более 2%. Внутрилабораторная воспроизводимость - устанавливает влияние случайных факторов на точность валидируемой методики. • Типичные исследуемые факторы: различные аналитики, различное оборудование, различные дни. • Например: испытание проводится двумя аналитиками в разные дни. Каждый специалист проводит 6 испытаний для каждого из 3-х контейнеров (флаконов) одной серии препарата. • Ход испытания – аналогично исследованию повторяемости. Находят: - Стандартное отклонение для определения каждым специалистом; - Среднее значение стандартного отклонения для определений двумя специалистами - Относительное стандартное отклонение (RSD) Критерий приемлемости для подтверждения внутрилабораторной воспроизводимости - величина относительного стандартного отклонения (RSD): отношение среднего значения стандартного отклонения к среднему значению активности (количественного содержания) по результатам определения двумя специалистами (%). • RSD – не более 3%. Межлабораторная воспроизводимость – оценивается путем проведения межлабораторных исследований. (Необязательна для генерических препаратов). Правильность Правильность МИ - выражение степени соответствия между принятым эталонным значением и значением, полученным на основании большой серии результатов испытаний по данной методике . ? Принятое эталонное значение – например, теоретически рассчитанное значение содержания вещества в модельном растворе ? Испытание проводят трехдозным методом как описано выше ? Правильность оценивают не менее чем для 9 определений для трех различных концентраций вещества (80%, 100%, 130% от номинального содержания в ЛС или АФИ): по три определения (по три чашки) для каждой концентрации. Например: • ЛС ванкомицин, порошок лиофилизированный 500 мг во флаконах содержит 500 мг активного вещества (ванкомицина гидрохлорида - АФИ). 80% - 400 мг – образец №1 100% - 500 мг – образец №2 130% - 650 мг – образец №3 ? Готовят соответствующие модельные растворы АФИ трех концентраций и проводят определение трехдозным методом диффузии в агар Анализ результатов Критерий приемлемости - величина процента восстановления для каждой концентрации: отношение экспериментально полученной концентрации к теоретически рассчитанной (в %) Процент восстановления должен быть в пределах 98 – 102% • Для образца №1 – 99% • Для образца №2 – 99,2% • Для образца №3 – 98,3% Подтверждена правильность МИ. Подтверждение прецизионности и правильности методики свидетельствуют о ее пригодности для контроля качества данного ЛС или АФИ. Вывод – данная методика гарантирует получение стабильных, достоверных результатов. Валидация методик определения микробиологической чистоты и стерильности Испытание на стерильность – подтверждение отсутствия в испытуемом образце жизнеспособных микроорганизмов Испытание на микробиологическую чистоту: • число бактерий и грибов – не более установленного критерия приемлемости • отдельные виды микроорганизмов не должны обнаруживаться в установленной массе (объеме) ЛС. Валидация микробиологических методов контроля (микробиологической чистоты и стерильности) - минимально cтатистически подтвержденная пригодность методики, которая проводится на трех различных сериях препарата с учетом всех возможных факторов влияния на достоверность результатов экспериментальных исследований (питательные среды, растворы, персонал). Поэтому валидационные исследования в отношении определения микробиологической чистоты и стерильности проводят: • на трех различных сериях препарата • трех разных партиях питательных сред • с привлечением трех специалистов, для исследования каждым из них по одной серии препарата • в разные дни (желательно). Методика определения качества лекарственных средств по микробиологическим показателям считывается валидной, если для трех различных серий препарата, на трех разных партиях питательных сред, у трех разных специалистов пригодность методики была подтверждена. Пригодность методики испытания: 1. Использование фармакопейных тест- микроорганизмов и подготовка их в соответствии с фармакопейными требованиями. 2. Подготовка образца - проводится аналогичным образом, как для испытаний микробиологической чистоты нестерильных лекарственных средств или стерильности. 3. Соблюдение условий приготовления питательных сред, проверка их ростовых, индикаторных и ингибирующих свойств (для каждой серии). 4. Проверка наличия/отсутствия антимикробного действия (среднее количество КОЕ на чашках с ЛС должно отличаться не более чем в 2 раза от количества КОЕ в контроле). 5. Нейтрализация антимикробного действия подходящим образом. 6. Предварительная оценка эффективности и безвредности инактиваторов и ПАВ. 7. Проведение контроля (рост культуры на питательной среде без ЛС) и отрицательного контроля (использование для инокуляции стерильного растворителя). 8. Демонстрация возможности обнаружения микроорганизмов- контаминантов в выбранных условиях проведения эксперимента. Все методики и испытания, включенные в фармакопеи сторон-участников ICH (Европейская Фармакопея, Фармакопея США и Фармакопея Японии), являются валидированными, и требуют только проведения верификации (проверки). Верификация должна подтвердить на основании экспериментальных данных, что данная лаборатория в состоянии корректно воспроизвести фармакопейную методику или испытание (т.е., для конкретного аналитического оборудования, для данных используемых реактивов, в данных условиях окружающей среды, при выполнении анализа аналитиками данной лаборатории и т.п.). Фармакопейные методики могут использоваться для контроля качества готовых лекарственных препаратов только после подтверждения, что конкретный состав не приводит к неприемлемому ухудшению правильности, линейности, или прецизионности методики. Нельзя без экспериментального подтверждения предполагать, что валидированная фармакопейная методика или испытание будут давать корректные результаты для лекарственного препарата с иным составом, чем тот, который использовался для валидации фармакопейных методик и испытаний [14,15]. РУКОВОДСТВО по валидации методик анализа лекарственных средств (методические рекомендациии http://doc.knigi-x.ru/22metodichka/283605-1-associaciya-rossiyskih-farmacevticheskih-proizvoditeley-rukovodstvo-validacii-metodik-analiza-lekarstvennih-sredstv-me.php Одно из наиболее важных изменений в руководстве GMP (Раздел 6) касаются верификации аналитических методик. Как известно, методики аналитических испытаний должны быть валидированы. В то же время фармакопейные методики по существу, не требуют валидации. В рамках ст. 1226 USP и новых требований GMP верификация применяется к фармакопейным (компендиальным) методикам при их использовании в реальных условиях в конкретной лаборатории. Она должна проводиться перед первым использованием по назначению, то есть для подтверждения того, что данная методика дает ожидаемые результаты в условиях конкретной лаборатории. Данной процедурой проверяются отдельные валидационные характеристики. Требование к объему верификации учитывает наличие опыта у персонала, сложность методики, используемого оборудования и анализируемого объекта. Это необходимо оценивать при определении объема верификации, то есть того, какие валидационные характеристики будут включены в протокол (специцифичность, прецизионность, линейность, воспроизводимость). В процессе верификации исследуются те валидационные характеристики, которые доказывают применимость методики, то есть наиболее ключевые, значимые. В соответствии со ст. 1226 USP ключевым параметром при верификации фармакопейной методики в применении ее к анализу субстанции и готового лекарственного средства является специфичность. То есть в протоколах верификации данный параметр необходимо использовать всегда. К примеру, специфичность хроматографических методик может верфицироваться тестом «Проверка пригодности» с требованием по коэффициенту разделения. Различные схемы синтеза субстанций (разные профили примесей) могут верифицироваться с помощью таких характеристик, как предел обнаружения примесей (или количественное определение) и прецизионность. Вследствие влияния различных вспомогательных веществ в готовых лекарственных средствах, антиоксидантов, буферных растворов, веществ, извлекаемых из материалов контейнеров, требуется верификация процента извлечения целевого компонента – аналита, то есть доказательство того, что он остается прежним или стабильным, как это указано в фармакопейной методике. Таким образом, рекомендуемые верификационные характеристики, согласно ст. 1226 USP, следующие: - при количественном определении примесей в субстанциях или готовых лекарственных средствах в протокол должны быть включены определение специфичности, предел количественного определения и прецизионность; - если используется вариант предельного определения примесей, следует учитывать 2 параметра: специфичность и пределе обнаружения; - если проводится количественное определение действующего вещества в субстанциях или готовых лекарственных средствах, необходимо исследовать специфичность, линейность и прецизионность (согласно рекомендациям эксперта Лювига Хьюбера) http://www.apteka.ua/article/325098 Верификация не требуется для простых фармакопейных методов, например, при определении потери в массе при высушивании, определении общей золы, кислотного числа, перекисного числа (волюметрические методы количественного определения), рН, то есть при использовании простых инструментальных методов. Согласно ст. 1058 USP это зависит также от сложности приборов. Для таких методов тем не менее необходимо учитывать специфику пробоподготовки (если при их подготовке аналит не теряется, верификация не требуется). Для определения простых инструментальных методов также можно воспользоваться пояснениями USP, а именно ст. 1058, в которой приводится классификация оборудования. Так, рекомендуемся делить оборудования на 3 категории – А, В, С и устанавливать для каждой группы соответствующий набор квалификационных тестов. На основании этого можно сказать, что методы, используемые с простыми приборами (группа А), являются простыми и не требуют верификации. В группу А включено вспомогательное оборудование: магнитные мешалки, центрифуги, ультразвуковые бани, то есть не измерительное оборудование. К группе В относятся приборы для точечного измерения – весы, приборы для измерения температуры плавления, титраторы, р\н-метры и пр. В группу С входит сложное оборудование, подлежащее полной квалификации, - спектрофотометры. хроматографы и др. Требования к планированию и документированию верификации не содержат нововведений. Они проводятся в соответствии с тем же порядком, что и при валидации (план, протокол, отчет, сравнение с критериями, анализ отклонений, финальный вывод в отношении успешности верификации). В случае если критерии приемлемости не выполняются и методика не пригодна для анализа данного объекта в конкретной лаборатории, требуется разработка альтернативной методики и ее полная валидация. По поводу опыта верификации методик скажу следующее: в соответствии с тербованиями 17025 мы методики проверяем на правильность и прецизионность, с точки зрения федеральной службы здравоохранения и соцразвития, фармакопейные методики на специфичность и так же на правильность и прецизионность. с форума http://www.anchem.ru/FORUM/read.asp?id=11237 http://www.vialek.ru/shop/catalog/books/detail/892/ Выводы ЖЕНЯ: Оставляем те параметры что мы и делаем – линейность, специфичность, прецизионность. 1. Требования к линейности А. из ОФС.1.1.0012.15 : Линейность методики – это наличие линейной зависимости аналитического сигнала от концентрации или количества определяемого вещества в анализируемой пробе в пределах аналитической области методики. При валидации методики ее линейность в аналитической области про-веряют экспериментально измерением аналитических сигналов для не менее чем 5 проб с различными количествами или концентрациями определяемого вещества. Экспериментальные данные обрабатывают методом наименьших квадратов с использованием линейной модели: y = b · x + a, где х – количество или концентрация определяемого вещества; y – величина отклика; b – угловой коэффициент; a – свободный член (ОФС «Статистическая обработка результатов химического эксперимента»). Должны быть рассчитаны и представлены величины b, a и коэффициент корреляции r. В большинстве случаев используют линейные зависимости, отвечающие условию 0,99, и только при анализе следовых количеств рассматривают линейные зависимости, для которых 0,9. В отдельных случаях возможность линейной аппроксимации экспериментальных данных обеспечивается лишь после их математического преобразования (например, логарифмирования). Для некоторых методик анализа, в основу которых в принципе не может быть положена линейная зависимость между экспериментальными данными, определение концентрации или количества вещества проводят с использованием нелинейных калибровочных графиков. При этом график зависимости аналитического сигнала от количества или концентрации определяемого вещества может быть аппроксимирован подходящей нелинейной функцией с использованием метода наименьших квадратов, что выполнимо при наличии соответствующего валидированного программного обеспечения. В. Из руководства по валидации методик 2. Линейность Линейная зависимость должна быть исследована в пределах диапазона применения аналитической методики. Она может быть подтверждена непо-средственно на активной субстанции (путем разбавления исходного раствора) и/или, для лекарственных препаратов, на модельных смесях с использованием соответствующей процедуры, что может быть исследовано при изучении диа-пазона применения. По полученным данным строят график зависимости сигнала как функции концентрации или количества определяемого компонента и визуально оцени-вают его линейность. Если линейная зависимость наблюдается, то результаты обрабатывают подходящим статистическим методом, например, методом наи-меньших квадратов. В некоторых случаях для получения линейности данные следует подвергнуть предварительному математическому преобразованию. Должны быть определены и представлены: коэффициент корреляции, точка пе-ресечения с осью ординат, тангенс угла наклона прямой и остаточная сумма квадратов отклонений, а также график со всеми экспериментальными данными. Для оценки линейности может понадобиться анализ отклонений эксперимен-тальных данных от прямой. Некоторые аналитические методики, например, иммуноаналитические, не показывают линейности ни при каких математических преобразованиях. В та-ких случаях аналитический отклик должен быть описан подходящей функцией концентрации анализируемого вещества в образце. Для подтверждения линейности рекомендуется использовать не менее 5 концентраций. Другие подходы должны быть обоснованы. В наш протокол в раздел линейность дополнить: - не менее 5 определений - добавить вывод: в выводе сравнить коэффициент корреляции. - добавить в табличку строчку «Отношение величины сигнала к концентрации» как в статье из журнала № 2 2016г. 2. Специфичность А. из ОФС.1.1.0012.15 Специфичность – это способность аналитической методики однозначно оценивать определяемое вещество в присутствии сопутствующих компонентов. Доказательство специфичности валидируемой методики обычно основывается на рассмотрении полученных с ее использованием данных анализа модельных смесей известного состава. Специфичность валидируемой методики может быть доказана также соответствующей статистической обработкой результатов анализов реальных объектов, выполненных с ее использованием и, параллельно, с использованием другой, заведомо специфичной, методики (методики, специфичность которой доказана). 1.1 Для методик испытаний на подлинностьВалидируемая методика (или совокупность методик) должна обеспечивать достоверную информацию о присутствии данного действующего вещества в субстанции или лекарственной форме при наличии в ее составе предусмотренных рецептурой компонентов, что подлежит экспериментальному подтверждению. Подлинность действующего вещества в фармацевтической субстанции или лекарственном препарате устанавливают в сравнении со стандартным образцом или по физико-химическим или химическим свойствам, не характерным для других компонентов. 1.2 Для методик количественного определения и испытания на примесиДля валидируемой методики количественного определения и испытаний на примеси применяют одинаковые подходы − должна быть оценена ее специфичность в отношении определяемого вещества, т. е. должно быть экспериментально подтверждено, что присутствие сопутствующих компонентов не влияет непредусмотренным образом на результат анализа. Допускается оценка специфичности валидируемой методики как путем анализа модельных смесей известного состава, содержащих определяемое вещество, так и путем сравнения результатов анализов реальных объектов, полученных одновременно с использованием валидируемой и другой, заведомо специфичной методики. Результаты соответствующих экспериментов должны быть статистически обработаны. Недостаток специфичности испытания может быть компенсирован другим (другими) дополнительным испытанием. При валидации методик, если это целесообразно, могут использоваться образцы лекарственных средств, подвергнутые, с целью накопления в них примесей, воздействию экстремальных условий (света, температуры, влажности) или химически модифицированные любым подходящим способом. Для хроматографических методик показывают разрешение между двумя наиболее близко элюирующимися веществами при соответствующих концентрациях. В. Из руководства по валидации методик: 1. Специфичность Исследование специфичности проводится при валидации испытаний на идентификацию, контроль примесей и количественное определение. Способ подтверждения специфичности зависит от задач, для решения которых предназначена аналитическая методика. Если методика недостаточно специфична, применяют сочетание двух или более аналитических методик для достижения необходимого уровня избирательности. 1.1. Идентификация Испытания на идентификацию должны обеспечивать возможность различать соединения близкого строения, которые могут присутствовать в образце совместно с определяемым компонентом. Избирательность методики может быть подтверждена получением положительных результатов (возможно, путем сравнения с известным стандартным образцом) для образцов, содержащих определяемый компонент, и отрицательных результатов, полученных для образцов, не содержащих его. Для подтверждения отсутствия ложноположительных результатов испытание на идентификацию может быть проверено для веществ, имеющих близкое строение или сопутствующих анализируемому веществу. Выбор веществ, потенциально мешающих проведению испытания, должен быть обоснован. 1.2. Количественное определение и испытания на примеси При валидации хроматографических методик для подтверждения специ-фичности должны использоваться характерные хроматограммы с указанием индивидуальных веществ. Аналогичный подход используют и для других ме-тодов разделения. Для хроматографических методик разрешение должно быть исследовано для соответствующих концентраций веществ. Для подтверждения специфично-сти может быть использовано разрешение двух наиболее близко элюирующихся веществ. В случае использования неспецифичного метода количественного опре-деления необходимо применять дополнительные аналитические методики и подтверждать специфичность всего комплекса методик. Например, если коли-чественное определение действующего вещества при выпуске проводится тит-риметрическим методом, то его можно дополнить соответствующим испытанием на примеси. Для количественного определения и для испытаний на примеси применяют одинаковые подходы, описанные ниже. 1.2.1. Образцы примесей имеются Для метода количественного определения необходимо подтвердить избирательность определения анализируемого вещества в присутствии примесей и/или вспомогательных веществ. Это можно сделать внесением в образец (активную субстанцию или готовый лекарственный препарат) примесей и/или вспомогательных веществ в соответствующей концентрации и последующего доказательства того, что это не отразилось на получаемом результате (путем сравнения результатов, полученных на исходном и загрязненном образцах). Для испытаний на чистоту подтверждение избирательности проводят путем загрязнения активной субстанции или готового лекарственного препарата соответствующими количествами примесей и доказательства разделения этих примесей как друг от друга, так и от других компонентов образца. 1.2.2. Образцы примесей отсутствуют Если образцы примесей или продуктов разложения отсутствуют, подтверждение специфичности проводят путем сравнения результатов анализа образцов, содержащих примеси или продукты разложения, полученных с помощью предлагаемой методики и другой арбитражной методики. В качестве последней может быть использована фармакопейная методика или другая валидированная методика. Этот подход предполагает предварительное загрязнение образца продуктами разложения путем выдерживания его в стрессовых условиях: воздействие света, тепла, влажности, гидролиз, окисление и т.п. При валидации методики количественного определения следует сравнить результаты анализов, полученных с использованием валидируемой и арбитражной методик. При валидации испытания на чистоту следует сравнить результаты определения примесей, полученные с использованием валидируемой и арбитражной методик. Для доказательства того, что пик анализируемого вещества соответствует только одному компоненту, используют тесты на чистоту пиков, например, с использованием диодно-матричного детектирования, массспектрометрии и др. Добавить вывод, а так можно оставить как есть, особо требований нет 3. Прецизионность А. из ОФС.1.1.0012.15 Прецизионность методики характеризуется рассеянием результатов, получаемых с ее использованием, относительно величины среднего результата. Мерой такого рассеяния является величина стандартного отклонения результата отдельного определения, полученная для выборки достаточно большого объема. Прецизионность оценивается для любой методики количественного определения по результатам не менее трех определений для каждого из трех уровней определяемых величин (нижнего, среднего и верхнего), лежащих в пределах аналитической области методики. Повторяемость также может оцениваться для любой методики количественного определения по результатам не менее шести определений для образцов с содержанием определяемого вещества, близким к номинальному. Во многих случаях оценка прецизионности может быть проведена по результатам обработки экспериментальных данных методом наименьших квадратов, как указано в ОФС «Статистическая обработка результатов химического эксперимента». Прецизионность должна исследоваться на однородных образцах и может оцениваться в трех вариантах: – как повторяемость (сходимость); – как внутрилабораторная (промежуточная) прецизионность; – как межлабораторная прецизионность (воспроизводимость). Результаты оценки методики анализа по каждому из вариантов прецизионности обычно характеризуются соответствующим значением величины стандартного отклонения результата отдельного определения. Обычно при разработке оригинальной методики определяется повторяемость (сходимость) результатов, получаемых с ее использованием. При необходимости включения разработанной методики в нормативную документацию дополнительно определяется ее внутрилабораторная (промежуточная) прецизионность. Межлабораторная прецизионность (воспроизводимость) методики оценивается при предполагаемом ее включении в проект общей фармакопейной статьи, фармакопейной статьи или в нормативную документацию на фармакопейные стандартные образцы. 7.1 Повторяемость (сходимость) Повторяемость аналитической методики оценивают по независимым результатам, полученным в одинаковых регламентированных условиях в одной лаборатории (один и тот же исполнитель, одно и то же оборудование, один и тот же набор реактивов) в пределах короткого промежутка времени. 7.2 Внутрилабораторная (промежуточная) прецизионность Внутрилабораторная (промежуточная) прецизионность валидируемой методики оценивается в условиях работы одной лаборатории (разные дни, разные исполнители, разное оборудование и т. д.). 7.3 Межлабораторная прецизионность (воспроизводимость) Межлабораторная прецизионность (воспроизводимость) валидируемой методики оценивается при проведении испытаний в разных лабораториях. В. Из Руководства по валидации методик: Валидационная характеристика прецизионность изучается для методик количественного определения и методик количественного определения примесей. 5.1. Сходимость Сходимость изучают, выполняя: a) не менее девяти определений, охватывающих диапазон применения методики (например, три концентрации / три определения для каждой) или b) не менее шести определений для образцов с содержанием анализируемого вещества, близким к номинальному. 5.2. Внутрилабораторная прецизионность Устанавливают влияние случайных факторов на прецизионность валидируемой аналитической методики. Типичными исследуемыми факторами являются различные дни, различные аналитики, различное оборудование и т.п. Не считается необходимым изучать влияние каждого фактора отдельно. При изучении влияния различных факторов предпочтительно использовать планирование эксперимента. 5.3. Воспроизводимость Воспроизводимость оценивают путем проведения межлабораторных исследований. Воспроизводимость должна быть изучена при стандартизации аналитической методики, например, при включении методики в фармакопею. Эти данные не включают в регистрационное досье. 5.4. Представление данных При изучении прецизионности следует представлять: стандартное отклонение, относительное стандартное отклонение и доверительный интервал. Добавить в табличку не менее шести определений для образцов с содержанием определяемого вещества, близким к номинальному. Добавить вывод В самом конце протокола написать заключение о том, что все норм ПРИМЕРЫ: Из руководства по валидации методик: 2.4. Пример проведения эксперимента и расчета критериев 2.4.1. Линейность, правильность и прецизионность Изучение сходимости и правильности рекомендуется проводить не менее чем из 9 определений, причем изучаемые концентрации должны охватывать диапазон методики. Поскольку изучение сходимости и правильности оценивается по отношению «найдено : введено» (в процентах) и проводится из данных, полученных при изучении линейности, наиболее оптимальной является схема, когда анализируют 9 модельных растворов, концентрации которых равномерно распределены в изучаемом диапазоне методики (плюс раствор сравнения, концентрация которого близка к номинальной). Ниже даны результаты расчета критических значений для параметров линейности, прецизионности и правильности для следующих испытаний (табл. 5): Количественное определение (субстанции и готовые лекарственные препараты), в зависимости от допусков содержания. Диапазон методики 80-120 %, изучаемые концентрации 80 %, 85 %, 90 %, 95 %, 100 %, 105 %, 110 %, 115 % и 120 % (шаг 5 %). Однородность содержания для дозированных лекарственных форм. Диапазон методики 70-130 %, изучаемые концентрации 70 %, 77.5 %, 85 %, 92.5 %, 100 %, 107.5 % 115 %, 122.5 % и 130 % (шаг 7.5 %). Растворение для твердых дозированных лекарственных форм. Диапазон методики дан для нормирования степени высвобождения не менее 75 % и не более 115 %, и соответственно составляет 55-135 %. Изучаемые концентрации 55 %, 65 %, 75 %, 85 %, 95 %, 105 %, 115 %, 125 % и 135 % (шаг 10 %). Таблица 5 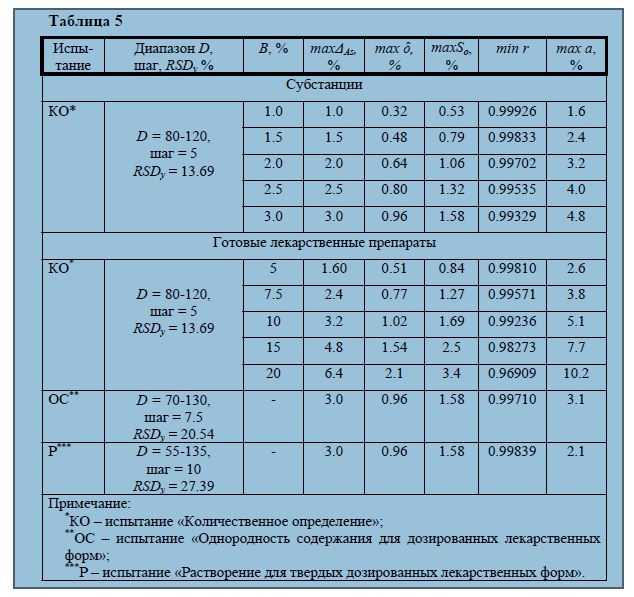 Для ЛС для человеков надо еще рассматривать ППХС |