Болезнь Канавалова. Определение Болезнь Вильсона (синонимы болезнь ВильсонаКоновалова, гепатолентикулярная дегенерация, гепатоцеребральная дистрофия) редкое наследственное заболевание,
 Скачать 140.43 Kb. Скачать 140.43 Kb.
|

|
Определение+ Болезнь Вильсона (синонимы: болезнь Вильсона-Коновалова, гепатолентикулярная дегенерация, гепатоцеребральная дистрофия) – редкое наследственное заболевание, связанное с нарушением метаболизма меди и избыточным ее накоплением в тканях и сочетанному поражению паренхиматозных органов (прежде всего печени) и головного мозга (преимущественно подкорковых ядер). История Первое описание клиники данного заболевания в 1912 году предложил американский невролог Сэмюель Вильсон, который отметил в ряде родословных некое фамильное заболевание, которое проявлялось в виде дистрофии в головном мозге, наличии цирроза печени и симптома "колец" ( бурого пигмента в роговице глаз описали Кайзер, в 1902; и Флейшер, в 1903году. ). Ученый назвал этот процесс «прогрессивная лентикулярная дегенерация» В качестве основных симптомов С. Вильсон выделил непроизвольные движения в конечностях и туловище, мышечная ригидность, а иногда были замечены психические расстройства. Ещё раньше Карл Вестфаль и Адольф Штрюмпелль описали патологию, которая имела клинические проявления сходные с рассеянным склерозом, она получила название «псевдосклероз». Заболевание характеризовалось непроизвольными движениями, повышением мышечного тонуса и психическими нарушениями вплоть до слабоумия. В дальнейшем оказалось, что прогрессивная лентикулярная дегенерация и псевдосклероз - это формы одного заболевания, которые Галль объединил в одно и назвал гепато-лентикулярной дегенерацией. Николай Васильевич Коновалов также изучал данное заболевание и предложил новое, более конкретное название процессу, такое как «гепато-церебральная дистрофия». Он расширил представление об этой патологии и выделил 5 форм, 4 из которых связаны с нервной системой, а 1 имела отношение к брюшной полости. Патогенез Основными ферментами, обеспечивающими транспорт меди в организме, являются АТФ-аза 7А и АТФ-аза 7В. мРНК АТФ-азы 7В обнаружена в гепатоцитах и капиллярах мозга. Этот фермент участвует в выведении меди организма (из крови в желчь) и из головного мозга в кровь. Именно его недостаток вызывает болезнь Вильсона Коновалова. Наиболее распространенной мутацией в гене АТР7В среди европеоидной расы является мутация 3207C>A (H1069Q) в экзоне 14. Генетически детерминируемое снижение функции медь-транспортирующей АТФ-азы в результате молекулярных дефектов в гене АТР7В приводит к снижению гепатобилиарной экскреции меди и нарушению встраивания меди в церулоплазмин, в результате экскретируется и циркулирует апоцерулоплазмин (ненагруженный медью, срок полувыведения которого сокращается вдвое. Повышение концентрации меди в печени приводит к некрозу гепатоцитов, воспалению, фиброзу, пролиферации желчных протоков и циррозу; в головном мозге – к некрозу нейронов с образованием полостей (кист). Изменения других органов и тканей, как правило, незначительны. Массивный выброс меди из гепатоцитов в кровь при их разрушении под воздействие какой-либо внешней причины (инфекция, интоксикация, ятрогенная реакция на тиоловые хелаты и др.) может привести к многократному повышению ее концентрации в плазме крови, а также к медьиндуцированному (на фоне гипоцерулоплазминемии) массивному гемолизу и далее к фульминантной печеночной несостоятельности. 4. Формы гепато-церебральной дистрофии: 1. Брюшная протекает в печени и имеет летальный исход. При этом более чувствительны являются дети до 7 лет. 2. Ранняя форма характеризуется форсированными проявлениями, которые так же, как и брюшная форма, поражает в основном детей (7-15 лет). Имеет следующие клинические проявления: бедность движений, мышечная ригидность, судорожный смех и плач, умеренное снижение интеллекта. Без лечения летальный исход наступает через 2–3 года. 3. Дрожательно-ригидная форма – это самая распространенная форма данного заболевания. Протекает медленно, но имеет внезапные ремиссии и ухудшения. Симптомами являются: субфебрильная температура, развитие тяжелой ригидности и ритмичного дрожания. Движение в основном отмечаются при волнении, а в покое утихают. Дрожательно-ригидная форма охватывает лиц 15 - 25 л., а продолжительность жизни сокращается до 6 лет. 4. Дрожательная форма отличается более поздним началом (20–30 лет) и продолжительным течением (10-15 и более лет). Клинически отмечаются дрожание, монотонная речь, угнетенная мимика, тяжелые изменения в психике и судорожные припадки. 5. Экстрапирамидно-корковая – наиболее редкая форма. Сопровождается судорожными припадками, слабоумием (т.к. происходит размягчение в коре головного мозга), длится от 6 до 8 лет и заканчивается летально. 5. Микроскопия В образцах печени пациента с болезнью Вильсона-Коновалова под микроскопом видно скопление гранул меди чёрно-зелёного цвета в пустых мембранах гепатоцитов 6. Клиническая картина Основными органами-мишенями при БВК являются печень, головной мозг и почки, так как заболевание является мультисистемным. Нарушение выведения меди из организма приводит к постепенному ее накоплению в органах и системах в определенной последовательности. После рождения ребенка с дефектным геном АТФ-азы 7В медь первоначально начинает накапливаться в печени. Поэтому у детей болезнь обычно манифестирует одним из вариантов поражения печени (абдоминальная форма), которое клинически проявляется в возрасте старше 4-5 лет. Печеночная манифестация является наиболее частой и отмечается у 40-50% больных. После того как печень насыщается медью, что в ряде случаев происходит бессимптомно, элемент перераспределяется системно, накапливаясь прежде всего в ЦНС, что ведет к нейропсихическим симптомам, которые чаще всего развиваются во 2-м и 3-м десятилетиях жизни. Неврологическая и психическая манифестации наблюдаются (соответственно) у 35 и 10% больных. У 15% больных заболевание проявляется гематологическими синдромами, прежде всего гемолитической анемией. В роговице накопление меди происходит после насыщения ею печени практически одновременно с появлением нейропсихической симптоматики. Смерть может наступить в результате отравления медью центральной нервной системы и на фоне нарушения функции печени. Если болезнь продолжается достаточно долго, то цирроз печени развивается всегда. 7. Диагностика Наиболее известным диагностическим симптомом ГЛД (болезни Вильсона-Коновалова) являются кольца Кайзера-Флейшера на радужной оболочке глаза. Они наблюдаются у 95% пациентов с церебральной и примерно у половины пациентов с абдоминальной формой заболевания. У детей элементы колец могут выглядеть в виде пигментных вкраплений или дуг вокруг радужки. Основными тестами, использующимися для диагностики заболевания, являются показатели обмена меди: церулоплазмин сыворотки, общая и свободная медь сыворотки, суточная экскреция меди с мочой, содержание меди в ткани печени. Для оценки прогноза выживаемости был разработан индекс БВ (Dhawan et al.), согласно которому оценка свыше 11 баллов связана с высокой вероятностью летального исхода при отсутствии срочной ортотопической трансплантации печени. 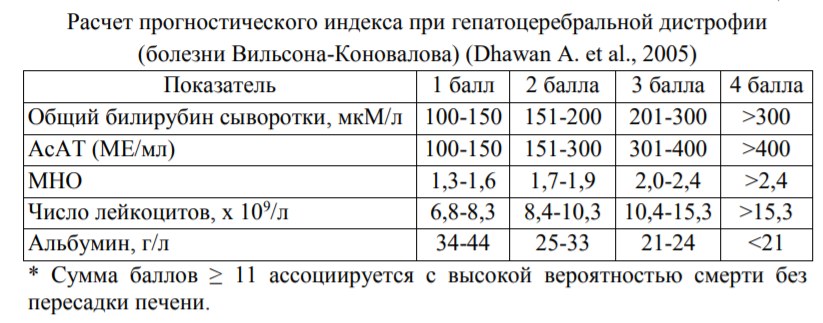 8. Лечение Основными методами лечения БВК является применение медьэлиминирующих препаратов, соблюдение строгой диеты со сниженным количеством меди в рационе и, при необходимости, проведение трансплантации печени. Целью терапии в бессимптомной стадии развития заболевания является предотвращение появления признаков заболевания и нормализация измененных лабораторных показателей; в стадии клинических проявлений – стабилизация и частичная регрессия основных симптомов заболевания, а также нормализация лабораторных показателей. |