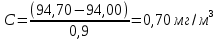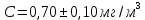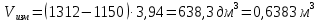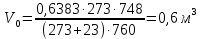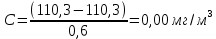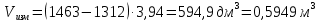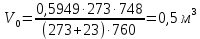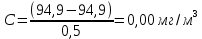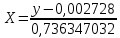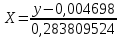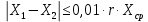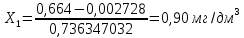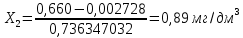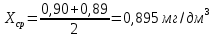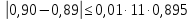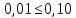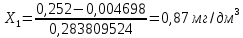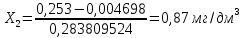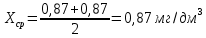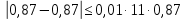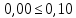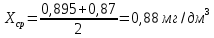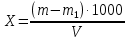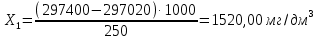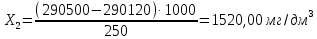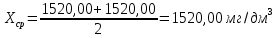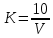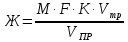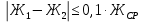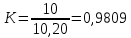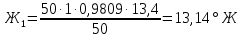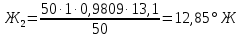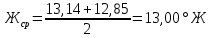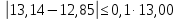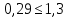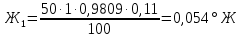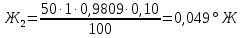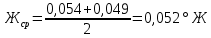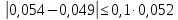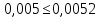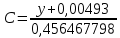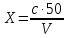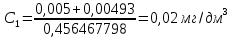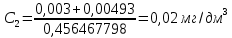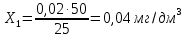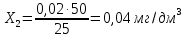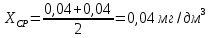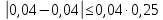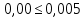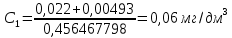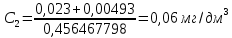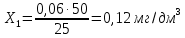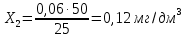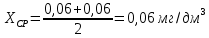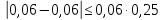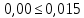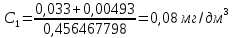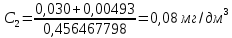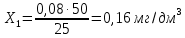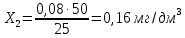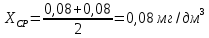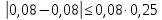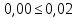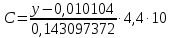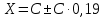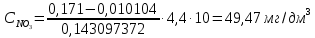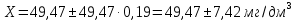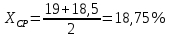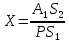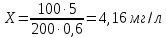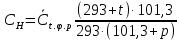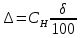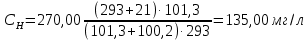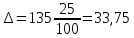Отчет по производственной практике. Содержание. 1. Общая характеристика предприятия 9 История предприятия 9
 Скачать 355.36 Kb. Скачать 355.36 Kb.
|

2 измерение
3 измерение
Максимально разовая концентрация по ПДК -0,5 мг/м3 Выводы: ознакомились с прибором для контроля качества воздуха (аспиратором ПУ-3Э), изучили метод определения массовой концентрации взвешенных веществ в атмосферном воздухе, освоили подготовку фильтров, определили содержание взвешенных веществ в воздухе рабочей зоны. Массовая концентрацию взвешенных веществ в анализируемой пробе атмосферного воздуха – 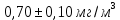 , что превышает установленную норму. , что превышает установленную норму.3.3. Определение массовой концентрации фосфат-ионов в питьевой воде фотометрическим методом Фотометрический метод определения массовой концентрации фосфат-ионов основан на их взаимодействии в кислой среде с молибдатом аммония и образованием фосфорно-молибденовой гетерополикислоты, которая восстанавливается аскорбиновой кислотой в присутствии сурьмяно-виннокислого калия до фосфорно-молибденового комплекса, окрашенного в голубой цвет. Максимум светопоглощения длине волны l = 690 нм. Цель работы: освоить методику определения фосфат-ионов в питьевой воде фотометрическим методом с молибдатом аммония. Определить содержание фосфат-ионов в анализируемой пробе. Приборы, посуда и реактивы: фотометр КФК-3 «ЗОМЗ», кюветы с толщиной поглощающего слоя 20 и 50 мм, весы лабораторные, ГСО состава раствора фосфат-ионов с массовой концентрацией 1 мг/см3, колбы мерные, пипетки мерные, цилиндры, воронки, колбы конические, стаканы для взвешивания, серная кислота, аммония молибдат, аскорбиновая кислота, калий сурьемно-виннокислый, калий марганцовокислый, сульфаминовая кислота, трилон Б. Порядок выполнения работы: Приготовление вспомогательных растворов Приготовление раствора молибдата аммония 3 г молибдата аммония помещают в стакан, растворяют в небольшом количестве дистиллированной воды, переносят в мерную колбу вместимостью 100 см3 и доводят до метки дистиллированной водой. В случае появления мути раствор следует отфильтровать. Раствор хранят в полиэтиленовой бутыли. Приготовление раствора аскорбиновой кислоты. 2,16 г аскорбиновой кислоты помещают в стакан, растворяют в небольшом количестве дистиллированной воды, переносят в мерную колбу вместимостью 100 см3 и доводят до метки дистиллированной водой. Раствор хранят в холодильнике в течение 3-х недель. Приготовление раствора антимонилтартрата калия. 0,34 г антимонилтартрата калия помещают в стакан, растворяют в небольшом количестве дистиллированной воды, переносят в мерную колбу вместимостью 500 см3 и доводят до метки дистиллированной водой. Приготовление раствора серной кислоты. В мерную колбу вместимостью 500 см3 наливают 400 см3 дистиллированной воды и осторожно приливают 70 см3 концентрированной серной кислоты. После охлаждения, раствор доводят до метки дистиллированной водой. Приготовление смешанного реактива. В колбе с притертой пробкой смешивают 125 см3 раствора серной кислоты, 50 см3 раствора молибдата аммония, 50 см3 раствора аскорбиновой кислоты и 25 см3 раствора антимонилтартрата калия. Смешанный реактив готовят непосредственно перед использованием. Ход анализа К 50 см3 пробы, профильтрованной на месте или в тот же день в лаборатории через плотный бумажный фильтр (синяя лента), или к меньшему объему, доведенному до 50 см3 дистиллированной водой, прибавляют 5,0 см3 смешанного реактива и через короткое время 0,5 см3 раствора аскорбиновой кислоты (как указано в п. 9.1.2 в присутствии некоторых мешающих веществ реактивы приливают в обратном порядке). Смесь перемешивают. Через 15 мин измеряют оптическую плотность полученного раствора при длине волны 690 нм по отношению к холостому раствору (холостой раствор готовится на дистиллированной воде с добавлением соответствующих реактивов). Содержание фосфат-ионов при l=50мм, рассчитывают по формуле:
где X – массовая концентрация форсфат-ионов, мг/дм3 y –оптическая плотность анализируемого раствора При l=20мм, формула приобретает вид:
Проводят два параллельных измерения и рассчитывают среднее значение при условии:
Расчет для анализируемой пробы питьевой воды Были проведены по 2 параллельных измерения для разбавленной пробы (при l=20мм) и неразбавленной пробы (при l=50мм) Оптическая плотность неразбавленных проб питьевой воды воды при толщине поглощающего слоя 50 мм: проба 1 - 0,664; проба 2 – 0,660 Содержание фосфат-ионов:
Оптическая плотность разбавленных проб питьевой воды воды при толщине поглощающего слоя 20 мм: проба 1 – 0,252; проба 2 – 0,253
Рассчитываем среднее значение между разбавленной и неразбавленной пробами анализируемой воды:
Выводы: освоили методику определения фосфат-ионов в питьевой воде фотометрическим методом с молибдатом аммония. Определили содержание фосфат-ионов в анализируемой пробе. Массовая концентрация фосфат ионов в анализируемой пробе воды – 0,88±0,12 мг/дм3. ПДК – 3,5 мг/дм3. Следовательно, анализируемая проба питьевой воды соответствует установленной норме. 3.4. Определение содержания сухого остатка в питьевой воде Величина сухого остатка характеризует общее содержание растворенных в воде нелетучих минеральных и частично органических соединений. Цель работы: освоить весовой метод определения содержания сухого остатка в питьевой воде, установить общее содержание растворенных нелетучих минеральных и частично органических соединений в анализируемой пробе. Приборы, посуда и реактивы: шкаф сушильный с терморегулятором, баня водяная, колбы мерные 250 мл, чашка фарфоровая выпарительная, эксикаторы. Порядок выполнения работы 250—500 см3 профильтрованной волы выпаривают в предварительно высушенной до постоянной массы фарфоровой чашке. Выпаривание ведут на водяной бане с дистиллированной водой. Затем чашку с сухим остатком помешают в термостат при 110 "С и сушат до постоянной массы. Обработка результатов Сухой остаток (X). мг/дм3, вычисляют по формуле:
где m — масса чашки с сухим остатком, мг; m2— масса пустой чашки, мг; V— объем воды, взятый для определения, см3 Данный метод определения сухого остатка дает несколько завышенные результаты вследствие гидролиза и гигроскопичности хлоридов магния и кальция и трудной отдачи кристаллизационной воды сульфатами кальция и магния. Эти недостатки устраняют прибавлением к выпариваемой воде химически чистого карбоната натрия. При этом хлориды, сульфаты кальция и магния переходят в безводные карбонаты, а из натриевых солей лишь сульфат натрия обладает кристаллизационной водой, но ее полностью удаляют высушиванием сухого остатка при 150—180 *С. Расхождения между результатами повторных определений не должны превышать 10 мг/дм3, если сухой остаток не превышает 500 мг/дм3; при более высоких концентрациях расхождение не должно превышать 2 отн. %. Расчеты для анализируемой пробы:
Выводы: освоили весовой метод определения содержания сухого остатка в питьевой воде, установили общее содержание растворенных нелетучих минеральных и частично органических соединений в анализируемой пробе. Величина сухого остатка в анализируемой пробе питьевой воды составила 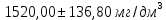 , что превышает установленную норму ПДК 1000,00мг/л. Анализируемая проба воды не соответствует требованиям. , что превышает установленную норму ПДК 1000,00мг/л. Анализируемая проба воды не соответствует требованиям.3.5. Определение жесткости питьевой воды Метод основан на образовании комплексных соединений трилона Б с ионами щелочноземельных элементов. Определение проводят титрованием пробы раствором трилона Б при pH = 10 в присутствии индикатора. Наименьшая определяемая жесткость воды — 0,1 °Ж. Цель работы: освоить комплексонометрический метод определения жесткости питьевой воды, проанализировать пробы воды до и после отчистки и сделать вывод о качестве отчистки. Приборы, посуда и реактивы: ГСО состава жесткости воды, весы лабораторные, рН-метр любого типа, мерные цилиндры, колбы мерные, пипетки градуированные, бюретка, колба плоскодонные, воронка лабораторная, стаканы химические, устройство для фильтрования, фильтры мембранные, шкаф сушильный, бумага универсальная индикаторная, вода дистиллированная, ГСО состава трилона Б, ГСО состава водного раствора ионов магния, стандарт-титр соляной кислоты, спирт этиловый, индикатор эриохром черный Т, аммония хлорид, аммиак хлорид, аммиак водный, кислота соляная, натрия гидроксид, натрия хлорид, натрия сульфид, гидроксиламина гидрохлорид. Порядок выполнения работы Установление коэффициента поправки к концентрации раствора трилона Б В коническую колбу вместимостью 250 см3 вносят 10,0 см3 раствора ионов магния (см. 4.3.2), добавляют 90 см3 бидистиллированной воды, 5 см3 буферного раствора (см. 4.3.3), от 5 до 7 капель раствора индикатора (см. 4.3.4.1) или от 0,05 до 0,1 г сухой смеси индикатора (см. 4.3.4.2) и сразу титруют раствором трилона Б (см. 4.3.1) до изменения окраски в эквивалентной точке от винно-красной (краснофиолетовой) до синей (с зеленоватым оттенком) при использовании индикатора эриохром черный Т, а при использовании индикатора хромовый темно-синий кислотный до синей (сине-фиолетовой). Раствор трилона Б в начале титрования добавляют довольно быстро при постоянном перемешивании. Затем, когда цвет раствора начинает меняться, раствор трилона Б добавляют медленно. Эквивалентной точки достигают при изменении окрашивания, когда цвет раствора перестает меняться при добавлении капель раствора трилона Б. Титрование проводят на фоне титрованной контрольной пробы. В качестве контрольной пробы можно использовать немного перетитрованную анализируемую пробу. За результат принимают среднеарифметическое значение результатов не менее двух определений. Значение коэффициента поправки должно быть равным 1,00 ± 0,03. Коэффициент поправки К к концентрации раствора трилона Б рассчитывают по формуле:
где V— объем раствора трилона Б, израсходованный на титрование, см3 10 – объем раствора ионов магния, см3 Порядок проведения определений Выполняют два определения, для чего пробу анализируемой воды делят на две части. В колбу вместимостью 250 см3 помещают первую часть аликвоты пробы анализируемой воды объемом 100 см3, 5 см3 буферного раствора, от 5 до 7 капель раствора индикатора или от 0,05 до 0,1 г сухой смеси индикатора и титруют раствором трилона Б. Вторую часть аликвоты пробы объемом 100 см3 помещают в колбу вместимостью 250 см3, добавляют 5 см3 буферного раствора, от 5 до 7 капель раствора индикатора или от 0,05 до 0,1 г сухой смеси индикатора, добавляют раствор трилона Б, которого берут на 0,5 см3 меньше, чем пошло на первое титрование, быстро и тщательно перемешивают и титруют (дотитровывают). Обработка результатов определения Жесткость воды Ж, °Ж, рассчитывают по формуле:
где М — коэффициент пересчета, равный 2Стр, где Стр — концентрация раствора трилона Б, моль/м3 (ммоль/дм3), (как правило М = 50); F — множитель разбавления исходной пробы воды при консервировании (как правило F= 1); К — коэффициент поправки к концентрации раствора трилона Б, рассчитанный по формуле (47); VТР — объем раствора трилона Б, израсходованный на титрование, см3; VПР— объем пробы воды, взятой для анализа, см3. За результат измерения принимают среднеарифметическое значение результатов двух определений. Приемлемость результатов определений оценивают исходя из условия:
Расчеты для анализируемых проб: Коэффициент поправки к концентрации раствора трилона Б
Анализируемая проба питьевой воды №1 (до очистки)
Анализируемая проба питьевой воды №4 (после очистки)
Выводы: освоили комплексонометрический метод определения жесткости питьевой воды, проанализировали пробы воды до и после отчистки. До отчистки жесткость воды составляет 13,00±1,95°Ж, при ПДК 10°Ж , после очистки менее 0,1 °Ж. Очистка проведена качественно. 3.6. Определение массовой концентрации общего железа с 2,2-дипиридилом Метод основан на взаимодействии ионов двухвалентного железа с 2,2-дипиридилом в области рН 3,5-8,5 с образованием окрашенного в красный цвет комплексного соединения. Интенсивность окраски пропорциональна массовой концентрации железа. Восстановление трехвалентного железа до двухвалентного проводится гидроксиламином. Окраска развивается быстро и устойчива в течение нескольких дней. Диапазон измерения массовой концентрации общего железа без разбавления пробы 0,05-2,00 мг/дм3. В этом интервале суммарная погрешность измерения с вероятностью Р=0,95 находится в пределах 0,01-0,03 мг/дм3 Цель работы: ознакомиться с методом измерения концентрации общего железа в питьевой воде, освоить приготовление растворов и определить концентрацию железа в анализируемых пробах. Приборы, посуда и реактивы: фотоколориметр КФК-3, кюветы с толщиной оптического слоя 2-5см, колбы мерные вместимостью 50,100 и 1000см3, пипетки мерные, аммоний уксуснокислый, гидроксиламин солянокислый, 2,2-дипиридил, квасцы железоаммонийные, кислота уксусная, спирт этиловый, вода дистиллированная. Порядок выполнения работы Для определения массовой концентрации общего железа исследуемую воду тщательно перемешивают и отбирают 25 см3 (или меньший объем, содержащий не более 0,1 мг железа) в мерную колбу вместимостью 50 см3. Прибавляют 1 см3 раствора гидроксиламина солянокислого, 2,00 см3 ацетатного буферного раствора, 1,00 см3 раствора 2,2-дипириднла и доводят до метки дистиллированной водой. После добавления каждого реактива содержимое колбы перемешивают. Раствор оставляют на 15-20 мин для полного развития окраски. Окрашенный раствор фотометрируют, применяя зеленый светофильтр (λ=540 нм) и кюветы с толщиной оптического слоя 2-5 см, по отношению к дистиллированной воде, в которую добавлены те же реактивы. Массовую концентрацию железа находят по градуировочному графику по формуле:
где y-оптическая плотность анализируемого раствора Массовую концентрацию общего железа вычисляют по формуле:
где с – концентрация железа, найденная по градуировочному графику, мг/дм3 V – объем воды, взятый для анализа, см3 50 – объем, до которого разбавлена проба, см3 За окончательный результат анализа принимают среднее арифметическое результатов двух параллельных измерений, допустимое расхождение между которыми не должно превышать 25 % при массовой концентрации железа на уровне предельно допустимой. Результат округляют до двух значащих цифр. Расчеты для анализируемых проб: Анализируемая проба питьевой воды №3
X=менее 0,05 мг/дм3 Анализируемая проба питьевой воды №7
X=0,06±0,018 мг/дм3 Анализируемая проба питьевой воды №8
X=0,08±0,024 мг/дм3 Выводы: ознакомились с методом измерения концентрации общего железа в питьевой воде, освоили приготовление растворов и определили концентрацию железа в анализируемых пробах. Массовая доля железа в анализируемых пробах воды: №3 - менее 0,05 мг/дм3, №7 - 0,06±0,018 мг/дм3, №8 - 0,08±0,024 мг/дм3. Все анализируемые пробы воды соответствуют норме ПДК-0,3 мг/л. 3.7. Определение содержания нитратов в пищевой воде с использованием салициловокислого натрия Сущность метода заключается во взаимодействии нитратов с салициловокислым натрием в сернокислой среде с образованием соли нитросалициловой кислоты, окрашенной в желтый цвет, и последующим фотометрическим определением и расчетом массовой концентрации нитратов в пробе исследуемой воды. Цель работы: изучить фотометрический метод определения содержания нитратов с использованием салициловокислого натрия (метод Д), освоить подготовку проб исследуемой воды, опеределить массовую концентрацию нитратов в анализируемой пробе. Приборы, посуда и реактивы: МСО состава водных растворов нитрат-ионов, обратный холодильник, квасцы алюмоаммонийные, калий азотнокислый, фенол красталлический, серебро сернокислое, аммиак водный, колбы мерные, кобальт хлористый, натрий салициловокислый с массовой долей 95%. Порядок выполнения работы Подготовка проб исследуемой воды Устранение мешающих влияний При значении мутности более 1,5 мг/дм3 и/или цветности воды более 20°—25° цветности к 150 см3 пробы исследуемой воды добавляют 3 см3 суспензии гидроокиси алюминия, пробу тщательно перемешивают и после отстаивания в течение нескольких минут осадок отфильтровывают, первую порцию фильтра отбрасывают. Хлориды при массовой концентрации более 200 мг/дм3 удаляют добавлением раствора сернокислого серебра к 100 см3 пробы исследуемой воды в количестве, эквивалентном содержанию хлорид-ионов. Осадок хлорида серебра отфильтровывают или отделяют центрифугированием. 10 см3 пробы исследуемой воды помещают в фарфоровую чашку, прибавляют 1 см3 раствора салициловокислого натрия и выпаривают на водяной бане досуха. После охлаждения сухой остаток увлажняют добавлением 1 см3 концентрированной серной кислоты, тщательно растирают его стеклянной палочкой и оставляют на 10 мин. Затем добавляют 5-10 см3 дистиллированной воды и количественно переносят в мерную колбу вместимостью 50 см3, прибавляют 7 см3 раствора гидроокиси натрия, доводят объем дистиллированной водой до метки и перемешивают. Далее в течение 10 мин после прибавления раствора гидроокиси натрия проводят определение, пока окраска пробы не изменилась. Измеряют оптическую плотность анализируемой пробы воды. В качестве холостой пробы используют дистиллированную воду. Массовую долю нитратов в воде рассчитывают по формуле:
где y-оптическая плотность анализируемого раствора Результат представляют в виде:
Расчет для анализируемой пробы:
Выводы: изучили фотометрический метод определения содержания нитратов с использованием салициловокислого натрия (метод Д), освоили подготовку проб исследуемой воды, опеределили массовую концентрацию нитратов в анализируемой пробе. Массовая концентрация ионов NO3 в анализируемой пробе воды составляет 49,47±7,42 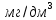 , что не превышает установленную норму ПДК=50 мг/дм3. , что не превышает установленную норму ПДК=50 мг/дм3. 3.8. Определение нитратов в продукции растениеводства Сущность метода состоит в извлечении нитратов из анализируемого материала раствором алюмокалиевых квасцов с последующим измерением их концентрации в полученной вытяжке с помощью ионоселективного электрода. Для ускорения анализа вместо вытяжки может быть использован сок анализируемой продукции, разбавленный раствором алюмокалиевых квасцов. При анализе капусты для разрушения примесей, мешающих определению нитратов, дополнительно проводят их окисление марганцевокислым калием. Цель работы: освоить ионометрический метод определения нитратов в продукции растениеводства, ознакомиться с правилами отбора проб и пробоподготовкой, определить количественное содержание нитратов в молодом картофеле и молодой капусте. Приборы, посуда и реактивы: преобразователь ионометрический и-500, ионоселективный нитратный электрод, электрод вспомогательный лабораторный хлорсеребряный, весы лабораторные, колбы мерные вместимостью 50, 100 и 1000 см3, дозатор вместимостью 50 см3, Пипетки вместимостью 1, 5, 10, 50 и 100 см3, стаканы лабораторные вместимостью 100, 200, 500 и 1000 см3,ступка фарфоровая с пестиком, мешалка лабораторная магнитная. пластмассовая терка, нож, стеклянные палочки, квасцы алюмокалиевые (KАI(SO)4·12Н2O, калий хлористый (KСI), калий азотнокислый (KNO3), калий марганцевокислый (KМnO4), кислота серная (Н2SO4), вода дистиллированная, перекись водорода, 33%-ный раствор. Порядок выполнения работы: Отбор проб для анализа Картофель. С участка площадью до 10 га по диагонали выкапывают без выбора не менее 20 кустов, расположенных через равные промежутки. Клубни отряхивают от земли и отделяют от столонов. Из каждого куста отбирают по одному среднему и крупному клубню. Капуста белокочанная, краснокочанная и цветная. Отбирают не менее 10 типичных кочанов, равномерно расположенных по диагонали, с каждого участка площадью 5 га. С кочанов снимают верхние кроющие листья. Общая масса объединенной пробы плодов должна составлять не менее 3 кг. Пробы к анализу готовят следующим образом: Картофель. Клубни моют водой, вытирают чистой тканью досуха и разрезают крестообразно вдоль оси "столон-ростовая часть" на 4 равные части. От каждого клубня берут четвертую часть, отобранный материал используют для анализа. Капуста. Кочаны разрезают крестообразно вдоль вертикальной оси на 4 или 8 равных частей и берут соответственно по 1/4 или 1/8 части от каждого кочана в пробу для анализа. При этом отбрасывают верхние несъедобные листья и остаток кочерыжки. Пробы необходимо готовить в количестве не менее двух, так как в случае повышенного содержания нитратов может возникнуть необходимость повторения анализа. Приготовление растворов сравнения Растворы сравнения азотнокислого калия готовят из основного раствора азотнокислого калия в день проведения анализа, используя для разбавления раствор алюмокалиевых квасцов, который применяется для анализа. Раствор сравнения с концентрацией СNO3=0,01 моль/дм3 (2). Основной раствор азотнокислого калия, разбавляют в 10 раз раствором алюмокалиевых квасцов. Для этого в мерную колбу вместимостью 100 см3 отбирают пипеткой 10 см3 основного раствора с 1, доводят до метки раствором алюмокалиевых квасцов и перемешивают. Раствор сравнения с концентрацией СNO3 =0,001 моль/дм3 (3). Раствор, приготовленный по предыдущему абзацу, разбавляют в 10 раз раствором алюмокалиевых квасцов. Раствор сравнения с концентрацией СNO3=0,0001 моль/дм3 (4). Предыдущий раствор, разбавляют в 10 раз paствором алюмокалиевых квасцов. Мембранный ионоселективный нитратный электрод и хлорсеребряный электрод сравнения готовят к работе в соответствии с инструкциями, прилагаемыми к ним. Проведение анализа Пробы измельчают с помощью терки. 10,0 г измельченного материала взвешивают с точностью до второго десятичного знака, помещают в стакан вместимостью около 100 см3, наливают 50 см3 1%-ного раствора алюмокалиевых квасцов и перемешивают с помощью мешалки в течение трех минут. При анализе капусты 10,0 г измельченного материала взвешивают с точностью до первого десятичного знака, помещают в стакан вместимостью 100 см3, наливают 50 см3 экстрагирующего раствора, перемешивают с помощью мешалки в течение трех минут. Затем при перемешивании добавляют по каплям (2-3 капли) 33%-ный раствор перекиси водорода до обесцвечивания раствора. В полученных суспензиях измеряют концентрацию нитрат-ионов. Перед проведением исследования производят градуировку прибора в соответствии с РЭ. Закончив градуировку прибора, электроды погружают в испытуемый раствор и снимают показания в единицах 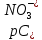 . Показания прибора считывают не ранее чем через 1 мин после прекращения дрейфа показаний прибора. Температура испытуемых проб и растворов сравнения должна быть одинаковой. Настройку прибора проверяют не менее трех раз в течение рабочего дня, используя каждый раз свежие порции растворов сравнения. . Показания прибора считывают не ранее чем через 1 мин после прекращения дрейфа показаний прибора. Температура испытуемых проб и растворов сравнения должна быть одинаковой. Настройку прибора проверяют не менее трех раз в течение рабочего дня, используя каждый раз свежие порции растворов сравнения.Полученные значения переводят в мг/кг 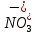 по таблицам. по таблицам.Расчеты для анализируемых проб пробы молодой картошки – 3,47, что соответствует 122 мг/кг. При норме до 250 мг/кг пробы молодой капусты – 2,82, что соответствует 554 мг/кг. При норме до 900 мг/кгВыводы: освоили ионометрический метод определения нитратов в продукции растениеводства, ознакомились с правилами отбора проб и пробоподготовкой, определили количественное содержание нитратов в молодом картофеле и молодой капусте. Оба анализируемый продуктов не превышают установленные нормы по содержанию нитратов. В пробе молодой картошки обнаружено 122 мг/кг нитратов, в молодой капусте – 554 мг/кг. 3.9. Определение жира в молочных продуктах кислотным методом Метод основан на выделении жира из молока, молочного напитка, молочных и молокосодержащих продуктов, кисломолочных продуктов, сыра и сырных продуктов, масла и масляной пасты, сливочно-растительного спреда и сливочно-растительной топленой смеси, мороженого под действием концентрированной серной кислоты и изоамилового спирта с последующим центрифугированием и измерении объема выделившегося жира в градуированной части жиромера. Цель работы: освоить методику определения жира в молочных продуктах кислотным методом, определить содержание жира в анализируемой пробе сметаны. Приборы, посуда и реактивы: жиромеры стеклянные, пробки резиновые для жиромеров, пипетки, груша резиновая, центрифуга лабораторная «Ока», баня водяная, штатив для жиромеров, термометр ртутный, весы лабораторные, цилиндр, ареометр общего назначения, секундомер, кислота серная, спирт изоамиловый, вода дистиллированная. Порядок выполнения работы В два жиромера типа 1-40 осторожно вхвешивают по 5 г сметаны с отсчетом до 0,005 г, тараясь не смочить горло, наливают дозатором по 10 см3 серной кислоты (плотностью от 1810 до 1820 кг/м3) Дозатором добавляют в жиромеры по 1 см изоамилового спирта. Уровень смеси в жиромере устанавливают на 1-2 мм ниже основания горловины жиромера, для чего разрешается добавлять несколько капель дистиллированной воды. Жиромеры закрывают сухими пробками, вводя их немного более чем наполовину в горловину жиромеров. Жиромеры встряхивают до полного растворения белковых веществ, переворачивая не менее 5 раз так, чтобы жидкости в них полностью перемешались. Рекомендуется для обеспечения проведения измерений наносить мел на поверхность пробок для укупорки жиромеров. Устанавливают жиромеры пробкой вниз на 5 мин в водяную баню при температуре (65±2)°С при частом встряхивании до полного растворения белка. Вынув из бани, жиромеры вставляют в стаканы центрифуги градуированной частью к центру. Жиромеры располагают симметрично, один против другого. При нечетном числе жиромеров в центрифугу помещают жиромер, наполненный водой вместо молока, серной кислотой и изоамиловым спиртом в том же соотношении, что и для анализа. Жиромеры центрифугируют 5 мин. Каждый жиромер вынимают из центрифуги и движением резиновой пробки регулируют столбик жира так, чтобы он находился в градуированной части жиромера. Жиромеры погружают пробками вниз на 5 мин в водяную баню при температуре (65±2)°С, при этом уровень воды в бане должен быть несколько выше уровня жира в жиромере. Жиромеры вынимают по одному из водяной бани и быстро производят отсчет жира. При отсчете жиромер держат вертикально, граница жира должна находиться на уровне глаз. Движением пробки устанавливают нижнюю границу столбика жира на нулевом или целом делении шкалы жиромера. От него отсчитывают число делений до нижней точки мениска столбика жира с точностью до наименьшего деления шкалы жиромера. Граница раздела жира и кислоты должна быть резкой, а столбик жира прозрачным. При наличии "кольца" (пробки) буроватого или темно-желтого цвета, различных примесей За результат измерений принимают среднеарифметическое значение результатов двух параллельных наблюдений, расхождение между которыми (сходимость) не превышает 0,5% Показания жиромера при измерениях в молоке, в т.ч. нежирном; кисломолочных продуктах, в т.ч. сметане, твороге; сливках (с массовой долей жира не более 40%), сливочном мороженом, пломбире, пахте и сыворотке соответствуют массовой доле жира в этих продуктах в процентах. Расчеты для анализируемой пробы: Было проведено исследование сметаны торговой марки «Иван Поддубный», 20% (АО Молочный комбинат «Воронежский») Проба 1 – показание жиромера: 19 Проба 2 – показание жиромера: 18,5
Выводы: освоили методику определения жира в молочных продуктах кислотным методом, определили содержание жира в анализируемой пробе сметаны. По результатам исследования сметана торговой марки «Иван Поддубный» не соответствует заявленному производителем содержанию жира (20%) и имеет массовую долю жира 18,75%. 3.10. Определение хлорорганических пестицидов (ГХЦГ-гамма (линдан)) в воде Метод основан на хроматографии хлорсодержащих пестицидов в тонком слое окиси алюминия, силикагеля или пластинок «Силуфол» в различных системах подвижных растворителей после экстракции их из исследуемых образцов и очистке экстрактов. Подвижным растворителем служит гексан или гексан в смеси с ацетоном. Места локализации препаратов обнаруживают после опрыскивания пластинок раствором аммиаката серебра с последующим ультрафиолетовым облучением или после облучения ультрафиолетовым светом пластинок «Силуфол», содержащих о-толидин. Цель работы: ознакомиться с методом тонкослойной хроматографии, освоить методику определения хлорорганических пестицидов в воде, определить содержание линдана в анализируемой пробе. Посуда, приборы и реактивы: Ацетон х.ч. Аммиак водный х.ч. Алюминия окись II степени активности для хроматографии (А) ч. Просеивают через сито 100 меш. Алюминия окись, пропитанная серной кислотой (Б). Две весовые части окиси алюминия (или окиси кремния) помещают в фарфоровую ступку, заливают одной объемной частью серной кислоты и тщательно перемешивают. Смесь готовят непосредственно перед подготовкой колонок для очистки экстрактов из проб шротов, жмыха, лузги. Бензол х.ч. Гексан ч. Калий щавелевокислый ч.д.а. Кальций сернокислый ч.д.а. Просушивают 6 ч в сушильном шкафу при 160 °С. Просеивают через сито 100 меш. Кремния окись для люминофоров ч. Натрий сернокислый безводный, ч. Натрий углекислый кислый, х.ч. Натрий хлористый х.ч., насыщенный раствор. Петролейный эфир (т. кип. 40 - 70 °С). Перекись водорода х.ч. (30 %-ный водный раствор). Проявляющий реактив № 1: 0,5 г азотнокислого серебра растворяют в 5 мл дистиллированной воды, прибавляют 7 мл аммиака и доводят объем раствора до 100 мл ацетоном; в готовый раствор добавляют 0,2 мл перекиси водорода. Раствор следует хранить в колбе с притертой пробкой в темном месте в течение 3 дней. На пластинку 9´12 см расходуется 8 - 10 мл раствора. Проявляющий реактив № 2: 0,5 г азотнокислого серебра растворяют в 5 мл дистиллированной воды, добавляют 10 мл 2-феноксиэтанола и доводят объем раствора до 200 мл ацетоном, затем добавляют 6 капель 30 % -ной перекиси водорода. Серебро азотнокислое ч.д.а. Серная кислота ч. Силикагель АСК (Воскресенского химкомбината). Силикагель КСК, просеянный через сито 100 меш. Стандартные образцы: ДДТ, ДДД, ДДЭ, альдрин, изомеры ГХЦГ, гептахлор, метоксихлор, кельтан, эфирсульфонат, дактал, тедион х.ч. Стандартные растворы: 10 мг соответствующего пестицида растворяют в мерной колбе на 100 мл в гексане и доводят до метки этим растворителем. Стандартные растворы необходимо хранить в стеклянной посуде с притертыми пробками в холодильнике. Стеклянная вата, очищенная концентрированной серной кислотой, промытая дистиллированной водой и высушенная, о-Толидин ч., 1 %-ный раствор в ацетоне. 2-феноксиэтанол. Этиловый спирт-ректификат. Хлороформ х.ч. Четыреххлористый углерод х.ч. Диэтиловый эфир (для наркоза). Натрий сернокислый, 2 %-ный водный раствор. Натрий сернокислый, насыщенный раствор. Баня водяная. Вакуумно-ротационный испаритель или прибор для отгонки растворителей. Воронки химические диаметром 6 см. Воронки делительные на 100, 250, 500 мл. Гомогенизатор или измельчитель тканей. Камера для опрыскивания. Камеры для хроматографирования размером 150´200, 105´165 мм. Колбы мерные на 50 и 100 мл. Колбы на шлифах емкостью 100, 250 и 500 мм. Колбы круглодонные на шлифах емкостью 150, 250 и 500 мл. Микропипетки (для нанесения стандартных растворов). Пипетки или шприцы для нанесения проб. Пипетки на 1, 5 и 10 мл. Прибор для встряхивания. Пластинки стеклянные 9´12, 13´18 см. Пульверизаторы стеклянные для опрыскивания пластинок. Сито на 100 меш (диаметр отверстий 0,147 мм). Стеклянные хроматографические колонки (диаметр´высота, мм) 20´400, 15´150. Ртутно-кварцевая лампа ПРК-4. Цилиндры мерные на 25, 50, 100, 250 и 500 мл. Чашки выпарительные № 3 и № 4. Порядок выполнения работы: Пластинки для хроматографии «Силуфол UV254» производства ЧССР перед использованием импрегнируют о-толидином. Для этого каждую пластинку погружают в 0,1 %-ный раствор о-толидина в ацетоне, налитого в камеру для хроматографирования. После того как фронт растворителя поднимется до верхнего края пластинки, ее вынимают и высушивают на воздухе, избегая прямого солнечного света, - пластинка готова к употреблению. Пластинки, импрегнированные о-толидином, хранят в эксикаторе. Пластинки «Силуфол UV254» производства ЧССР предварительно промывают дистиллированной водой в хроматографической камере, высушивают на воздухе и непосредственно перед использованием активируют в сушильном шкафу при температуре 65 °С в течение 4 мин. В рабочую пробу вносят 5 мкг ГСО С=100мкг/см3 (ГСО 8890-2007) Пробу 200 мл помещают в делительную воронку и экстрагируют пестициды, встряхивая в течение 3 мин, гексаном или петролейным эфиром тремя порциями по 30 мл, или диэтиловым эфиром тремя порциями по 50 мл. В объединенные экстракты насыпают 10 г безводного сернокислого натрия или фильтруют через воронку, заполненную на 2/3 сернокислым натрием. Экстракты переносят в прибор для отгонки растворителей и отгоняют растворитель до объема 0,2 - 0,3 мл. В случае необходимости экстракт чистят серной кислотой. На хроматографическую пластинку на расстоянии 1,5 см от ее края шприцем или пипеткой наносят исследуемую пробу в одну точку так, чтобы диаметр пятна не превышал 1 см. Остаток экстракта в колбочке смывают тремя порциями (по 0,2 мл) диэтилового эфира, которые наносят в центр первого пятна. Справа и слева от пробы на расстоянии 2 см наносят стандартные растворы, содержащие 10, 5 и 1 мкг исследуемых препаратов (или другие количества, близкие к определяемым концентрациям препаратов). Пластинки с нанесенными растворами помещают в камеру для хроматографирования, на дно которой за 30 мин до начала хроматографирования наливают подвижный растворитель. При использовании пластинок с тонким слоем окиси алюминия или силикагеля в качестве подвижного растворителя применяют гексан или смесь гексана с ацетоном (6:1) для препаратов, у которых величина Rf в гексане ниже 0,3. При использовании пластинок «Силуфол» подвижный растворитель - 1 %-ный раствор ацетона в гексане, а на пластинках «Силуфол», импрегнированных о-толидином, - гексан с диэтиловым эфиром (49:1). Край пластинки с нанесенными растворами может быть погружен в подвижный растворитель не более чем на 0,5 см. После того как фронт растворителя поднимется на 10 см, пластинку вынимают из камеры и оставляют на несколько минут для испарения растворителя. Далее пластинку орошают проявляющим реактивом и подвергают действию ультрафиолетового света в течение 10 - 15 мин (лампа ПРК-4). Пластинки следует располагать на расстоянии 20 см от источника света. При наличии хлорорганических пестицидов на пластинке появляются пятна серо-черного цвета. При использовании для анализа пластинок «Силуфол», импрегнированных о-толидином, их непосредственно после хроматографирования подвергают облучению ультрафиолетовым светом в течение нескольких минут. При наличии хлорорганических пестицидов в этом случае проявляются пятна сине-голубого цвета. Количественное определение осуществляют сравнением площадей пятен пробы и стандартных растворов. Между количеством препарата в пробе, не превышающим 20 мкг, и площадью его пятна на пластинке существует прямая пропорциональная зависимость. При большем содержании препарата следует использовать пропорциональную часть исследуемого экстракта. Количество препарата в пробе (X, мг/кг или мг/л) вычисляют по формуле:
где А1 - содержание препарата в стандартном растворе, мкг; S1 - площадь пятна стандартного раствора, мм2; S2 - площадь пятна пробы, мм2; Р - масса или объем исследуемой пробы, г или мл. Расчет для анализируемой пробы: При добавлние к рабочей пробе 5 мкг ГСО:
Без добавления ГСО в рабочей пробе хлорорганического пестицида не обнаружено. Погрешность измерения – 50% Выводы: ознакомилиись с методом тонкослойной хроматографии, освоили методику определения хлорорганических пестицидов в воде, определить содержание линдана в анализируемой пробе с добавлением ГСО – 4,16±2,08 мг/л 3.11. Измерение концентрации вредных веществ индикаторными трубками в воздухе рабочей зоны Сущность метода заключается в изменении окраски индикаторного порошка в результате реакции с вредным веществом (газом или паром) в анализируемом воздухе, просасываемом через трубку. Измерение концентрации вредного вещества производится по длине изменившего первоначальную окраску слоя индикаторного порошка в трубке (линейно-колористическая индикаторная трубка) или по его интенсивности (колориметрическая индикаторная трубка). Цель работы: освоить метод измерения концентрации вредных веществ индикаторными трубками, определить массовую концентрацию ацетона в воздухе рабочей зоны. Приборы, посуда и реактивы: индикаторные трубки, фильтрующие трубки, воздухозаборное устройство(аспиратор сильфонный АМ-5) Порядок проведения работы: Измерение концентраций вредных веществ в воздухе рабочей зоны проводят при следующих параметрах: барометрическое давление - от 90 до 104 кПа (680-780 мм рт.ст); относительная влажность - 30-80%; температура - от 288 до 303 К. Допускается отклонение от указанных параметров, если это предусмотрено в нормативно-технической документации на средства измерения. Выполнение измерений: Вскрыть индикаторную трубку модели ИТ-C3H6O/10,0 и трубку фильтрующую модели ТФ-C3H6O с обоих концов. Прокачать через ТФ 200 см3 анализируемого воздуха. Извлечь ТФ из аспиратора. Соединить ТФ с ИТ через переходник. Вставить ИТ в гнездо аспиратора АМ-5 концом, на который указывает стрелка, при необходимости использовать переходник прокачать необходимый объем 100 или 500 см3 анализируемого воздуха или газовой смеси через ИТ. В присутствие ацетона индикаторный порошок изменяет цвет с сине-зеленого на желтый. Определить концентрацию ацетона по шкале на ИТ или коробке, где «H» - точка совмещения начала индикаторного слоя с началом шкалы. При размытой или неровной границе раздела окрасок за результат измерения принимаю концентрацию, соответствующую усредненному значению наибольшей и наименьшей длины прореагировавшего участка индикаторного слоя. Для полуKчения достоверных результатов при выполнение измерения, использование ТФ является обязательным. Одну ТФ можно использовать не более чем с одной ИТ. Измерение концентраций вредных веществ, производят последовательно при производственных условиях. При этом используют количество индикаторных трубок, указанное в соответствующей нормативно-технической документации. За результат измерения принимают среднеарифметическое из 9 последовательных наблюдений. Результат измерения концентрации вредного вещества приводят к нормальным условиям (CН): температура 293 К, атмосферное давление 101,3 кПа (760 мм рт.ст.), относительная влажность 60% по формуле:
где 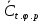 - результат измерения концентрации вредного вещества, при температуре окружающего воздуха t°С, относительной влажности φ % и атмосферном давлении p кПа, мг/м3; - результат измерения концентрации вредного вещества, при температуре окружающего воздуха t°С, относительной влажности φ % и атмосферном давлении p кПа, мг/м3;Относительная погрешность измерения не должна превышать ±35% в диапазоне до 2,0 предельно допустимых концентраций (ПДК) включительно и ±25% при концентрациях выше 2,0 ПДК при условиях. ПДК=800 мг/м3 Результат измерения представляют в виде: 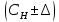 мг/м3 при доверительной вероятности 0,95. мг/м3 при доверительной вероятности 0,95. Величину абсолютной погрешности вычисляют по формуле:
В диапазоне до 1,0 ПДК включительно допускается увеличение погрешности до ±60%. Это значение относительной погрешности должно быть указано в нормативно-технической документации на средства измерения. Расчеты для проводимых измерений: Климатические условия помещения при отборе проб: атмосферное давление – 753 мм.рт.ст.=100,2 кПа температура воздуха - +21℃; относительная влажность – 75% 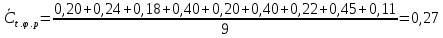 0,27 г/м3=270,00 мг/л
Выводы: освоили метод измерения концентрации вредных веществ индикаторными трубками, определили массовую концентрацию ацетона в воздухе рабочей зоны. Содержание ацетона в воздухе рабочей зоны – 135,00±33,75 мг/л. Воздух соответствует норме ПДК 3.12. Определение удельной эффективной активности естественных радионуклидов в строительных материалах Обнаружение гамма - излучения основано на регистрации эффектов, возникающих при его взаимодействии с веществом. Гамма - кванты, испускаемые атомными ядрами при радиоактивных превращениях, имеют определенные физические характеристики, которые можно использовать для их регистрации. Измеряя энергию и интенсивность испускаемых гамма - квантов, а также оценивая период полураспада их отдельных моноэнергетических групп, можно идентифицировать радионуклиды в измеряемых счетных образцах и достаточно точно определить их абсолютную активность. Цель работы: освоить лабораторный метод определения удельной эффективной активности естественных радионуклидов в строительных материалах, определить удельную эффективную активность радионуклидов в анализируемой пробе щебня из горных пород, установить класс строительного материала (изделия). Порядок выполнения работы: Определение удельных активностей ЕРН в сыпучих материалах проводят на навесках, отобранных из представительной пробы. Представительную пробу получают путем перемешивания и квартования не менее 10 точечных проб, отобранных из контрольных точек. Отбор проб производят в соответствии с требованиями действующих нормативных документов. Представительную пробу с размером зерен более 5 мм измельчают до размеров зерен менее 5 мм. В зависимости от объема применяемого в радиометрической установке контейнера пробу массой от 2,5 до 10 кг упаковывают в двойной мешок, между стенками которого помещают паспорт пробы с наименованием материала, адреса предприятия, направившего пробу, места и даты отбора пробы. Контейнеры с навесками последовательно устанавливают в радиометрическую установку и проводят измерения в соответствии с методикой выполнения измерений. В качестве результатов измерений удельных активностей ЕРН в представительной пробе принимают средние арифметические значения удельных активностей каждого радионуклида по пяти навескам. Принятие решения об использовании строительных материалов согласно гигиеническим нормативам производится по таблице
Расчет для анализируемой пробы:
Масса пробы – 500г Измерение:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||