|
|
1 Основные исторические вехи в развитии биоинформатики
31. Сходство последовательностей биополимеров и эволюция. Основные понятия, связанные с реконструкцией эволюции методами биоинформатики. При анализе сиквенсов ДНК для отдельных генов путем множественного выравнивания установлено, что сходство нуклеотидов достигает более 50%(около половины нуклеотидов сходны в белок-кодир-й части гена цитохрома С у дрожжей, пшеницы и приматов. Вероятность, что такое сходство сиквенсов случайно, крайне мала. Источник подобия – общее происхожд-е, история эволюции организмов записана в их геномах. Основная задача вычислительной эволюц-й биологии – реконструир-ть эволюцию организмов и макс-но согласовать эволюцию с классиф-й. Элементарные эволюц-е явления, которые можно вычленить на нуклеотидном уровне, это транзиции, трансверзии, вставки и делеции, но главнейшими вопросами молекуляр-й эволюции остается скорость таких изменений и до какой степени родство связано с различиями в сиквенсах(Не зная скорости, мы не можем точно сказать, как давно произошла дивергенция). Главное противоречие, которое встреч-т молекуляр-я филогенетика: организмы, сходные по отдельным сиквенсам, оказ-ся очень различ-ми на уровне фенотипа, в особенности макропризнаков. Классификации, построенные на молекуляр-х признаках, и на морфологич-х, сильно различ-ся. Классификация – разнесение разнообр-я орг-го мира по неким классам, при котором не обязательно приним-ся в расчет истор-е происхож-е объектов. Филогения – путь историч-го развития той или иной классификац-й группы, включающий вымерших общих или раз-х предков. Основные понятия, связанные с реконструкцией эволюции.Гомология – сходство в связи с происхож-м от общего предка. Подобие – наблюдаемое сходство, согласно данным, собранным в наст-й момент времени, и не включ-х каких-либо истор-х гипотез. Кластеризация – сведение вместе сходных сиквенсов и др объектов в группы, внутри которых они более сходны между собой, чем с любыми др объектами вне кластера. Иерархическая кластеризация – многоступенч-е объединение мелких кластеров в более крупные. Филогения– описание топологии истор-х взаимосвязей между организмов, основанных на предполаг-й гомологии по набору признаков и/или на модели эволюц-х процессов.
32. Филогенетические деревья и их элементы. Внешняя группа и укоренение дерева. Фенетический и эволюционный подход к построению филогенетических деревьев. Основные понятия, связанные с реконструкцией эволюции на основе молекулярных данных (сиквенсов). Гомология – сходство в связи с происхождением от общего предка, т.е. основанное на исторической гипотезе. Подобие – наблюдаемое сходство, согласно данным, собранным в теперешний момент времени, и не включающих каких-либо исторических гипотез. Кластеризация – сведение вместе сходных сиквенсов и других объектов в группы (кластеры), внутри которых они более сходны между собой, чем с любыми другими объектами вне кластера. Иерархическая кластеризация – многоступенчатое объединение мелких кластеров в более крупные. Филогения– описание топологии исторических взаимосвязей между организмов, основанных на предполагаемой гомологии по набору признаков и/или на модели эволюционных процессов. Топология исторических взаимосвязей отображается с помощью филогенетического дерева. Основой для реконструкции эволюции являются гены, причем как экзоны, так и интроны внутри них. При этом по количеству несинонимичных замен кодонов можно судить о селективности или неселективности изменений на уровне нуклеотидов. Для изучения эволюционных связей между отдаленными таксонами берут консервативные, медленно изменяющиеся сиквенсы, а для близкородственных (например, виды одного рода или роды одного семейства) – быстро изменяющиеся сиквенсы. Почти неизменные гены не подходят для оценки эволюционного расстояния. Сильно разошедшиеся гены также не подходят, так как трудно поддаются выравниванию. Например, митохондриальный геном изменяется быстро, и полезен для изучения эволюции близкородственных видов. Гены рРНК консервативны, и на их основе доказана самостоятельность трех империй: архебактерий, бактерий, эукариот.
В филогенетическом исследовании выявляется родство и генеалогия видов, популяций, индивидов или генов. Наблюдаемые в настоящий момент группы, для которых мы реконструируем филогению, называются рабочими, или операционными таксономическими единицами (operational taxonomic units – OTU / ОТЕ). В качестве OTU выступают упомянутые выше объекты.
Реальные предки используются в молекулярной филогении очень редко из-за плохой сохранности ископаемой ДНК. Хотя некоторые сиквенсы ДНК неандертальца получены из костей, датированных возрастом 30 000 лет. Как правило, ископаемая ДНК содержит слишком много повреждений или пробелов, чтобы произвести выравнивание. Филогенетические деревья состоят из следующих элементов: узлов (node, vertex) и ветвей (ребер, клад – internodes, edges). Узлы делятся на конечные (сами ОТЕ), внутренние (между конечными и корневым) и у укорененного дерева имеется корневой узел. Внутрениие узлы представляют ненаблюдаемые (гипотетические) таксоны или предков (ancestors).
Филогенетические деревья бывают трех видов: кладограмма – древовидная схема, отображающая только генеалогию OTU, и ветви которой расположены в виде треугольных вилок; фенограмма (дендрограмма, кластерная диаграмма) – древовидная схема, отображающая и генеалогию, и отношения сходства OTU, и ветви которой расположены в виде прямоугольных вилок; филограмма (эволюционное дерево) – отображающие не только генеалогию, но и число элементарных эволюционных изменений, приведших к возникновению той или иной OTU, или же время отделения от предка (степень дивергенции), что отображается через длину ветвей. Дерево, показывающее всех потомков от одного предка, называется укорененным. Дерево, показывающее родство (происхождение), но не отображающее исторический ход событий, называется неукорененным. Филограммы различаются по графическому способу представления: прямоугольные; полярные; радиальные. Длина пути через филограмму – это сумма длин ребер при движении от предка A к наблюдаемой ныне единице B.
Для филограмм предположение, что различия между ныне живущими таксонами отражает степень их дивергенции верно только в том случае, когда скорости эволюции одинаковы для всех ветвей дерева. Однако известно много исключений,показывающих разные скорости эволюции между группами организмов. Укоренение обычно производят введением в анализ внешней группы (outgroup) – таксона, обособленность которого от внутренней группы (ingroup) не вызывает сомнений. Внешняя группа позволяет оценить скорости эволюции. Если внутренней группой будут приматы, а внешней – корова, и скорость эволюции у приматов постоянная, то ожидаются примерно одинаковые расстояния между видами приматов и коровой.
Два подхода, используемые для построения филогенетического дерева: фенетический – пренебрегает исторической моделью родства между ОТЕ и опирается только на чисто математическое расстояние между видами (как дистанция между векторами матрицы, согласно мере сходства и различия); основная процедура построения дерева – иерархическая кластеризация; таким образом, фенетика основана на простом сходстве;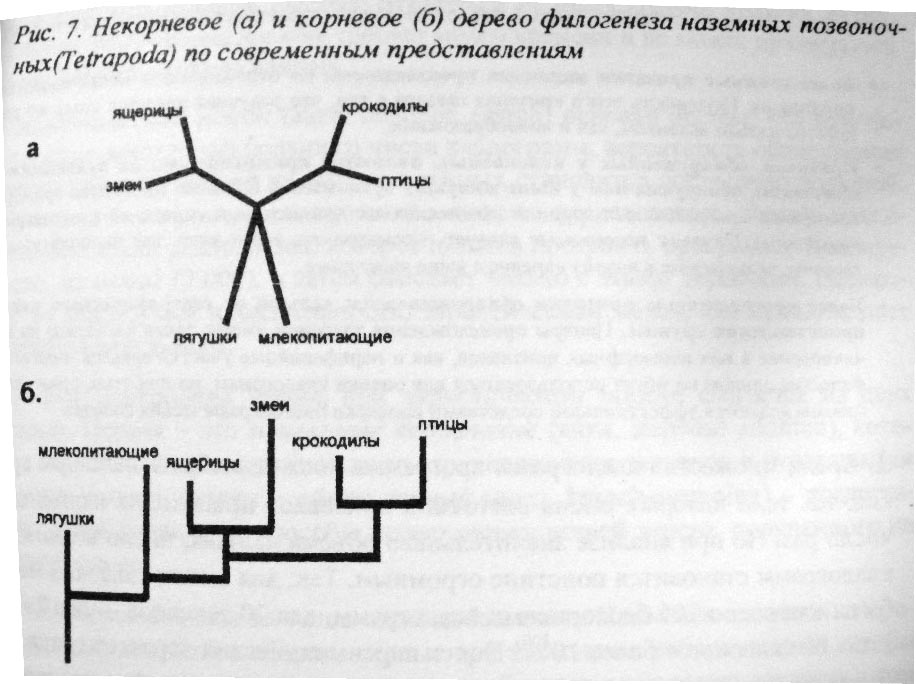
эволюционный (кладистика) – рассмотрение возможных путей эволюции и выбор оптимального дерева в соответствии с моделью эволюции; таким образом, кладистика основана на наиболее вероятной генеалогии.
Основные проблемы при реконструкции филогении на основе сиквенсов таковы: (1) множество вариантов сходства между сиквенсами, которые могут не быть статистически значимыми; (2) разная скорость эволюции в группах организмов; (3) если можно точно установить родство, может быть невозможно установить порядок, в котором таксоны разделились.
33. Основные геномные последовательности, используемые для установления родства между организмами и реконструкции эволюции.
Осн. задача вычислит-й эволюц-й биологии – реконструир-ть эволюцию организ-в и макс. согласовать эволюцию с классифик-й. Основой для реконстр-ции эволюции явл-ся гены..Части генома, используем. для установл.филогенет-городства.Гены, кодирующ. рРНК (рДНК)-располаг-ся в виде тандемных скоплений на определ-х хромос-х.
5’ (*) (*) 3’
-
NTS
|
ETS
|
185
|
ITS
|
5.8S
|
ITS2
|
285
|
NTS
|
** **
Кластер генов р-РНК.NTS-нетранскрибируемый спейсер, ETS-внешний транскрибир-й спейсер, ITS- внутр.транскрибир-й спейсер. Спейсеры-интроны.др.-экзоны. Такая стр-расвойст-на эукар-м. *-для генов дальнеродствен-х таксонов, **-для близкородств-х таксонов.Рибосомысост-т из 2-х отличающ-ся размерами субчастиц.Связь между ними слаба: при измен. параметров среды, ведущему к электростатич.дезэкраниров-ю фосфатных групп рРНК (пример: при сниж.конц-ции ионов Mg) риб-мадиссоц-т на субчастицы,привосстановл-и парам-в среды субчастицыреассоциир-т в исх.риб-мы.Отнош-е масс субчастиц
2:1; массы выраж-ся в измеряемых константах седиментации (скорость осажд-я в единицах Сведберга, S) при ультрацентрифуговании.Этот параметр в основе номенкл-ры рРНК,рибосом и рибосомных субчастиц. 70S риб-ма прокар-т сост-т из большой 50S субъед-цы (построен. на основе 2-х мол-л рРНК — 5S и 23S) и малой 30S субъед-цы (построен. на основе 16S рРНК). 80S рибосома эукариот сос-т из большой 60S субъед-цы (построен. на основе 3-х мол-л рРНК — 5S, 5,8S и 28S) и малой 40S субъед-цы (построен.на основе 18S рРНК).У млекопит-х сущ-т рибосомы цитоплазмы и митохонд-й. Митохондр. рибосомы: 12S и 16S.Гены цитохромов:–гемопротеины, связанные с мембраной и участв-т в электрич. транспорте.Есть 3 группы цитохровов: a,b,c. Цитохром с наиб. важен в генетич. кодировании. Процентное единообразие цитохрома с среди различных существ:чел-к-мышь-91,3%,чел-к-кукуруза-66,7%,чел-к-инфузория- 47,5%.Полные геномы исп-ся редко для филогении. Исп-ся геном Е. Coli.Ген цитохром с оксидазы: Фермент цитохром с-оксидазы явл-ся большим трансмембран-м белковым комплексом.Исп-ся для реконструкции филогении в животном мире COI – субъединиц I.Гены α и β тубулина: тубулинотн-ся к сем-ву глобулин-х белков. α и β тубулиныформир-т микротруб-ки, кот.присутст-т у всех эукариот-х кл-к. SINE и LINE относ-ся к сем-вуретротранспозонов.Отн-ся к недлин. повторам, применяем. у эукар-т.LINE имеет 2 рамки считывания, кодирующ.ф-циитранскрипции,кодир-т обрат. транскриптазу и эндонуклеазу. SINE–неавтономные,короткие и не кодирующ. все ф-ции, необход. для ретротранспозиции.Использов-е SINE и LINE для установл-я филогенетич-го родства.Короткие и длинные распределен. ядерные элем-ты– повторяющ-ся некодирующие последоват-ти, образ-т как мин. 30% генома чел-ка и более 50% генома нек-х высших раст-й.Особен-ти SINE, кот. позво-т использ-ть их как молекуляр. филогенетич. маркеры:1) SINE s либо присутс-т, либо отсутс-т в данном локусе генома;2) они случ. образом вставлены в некодирующ. части генома; появл. похожего SINE в одном и том же локусе указ-т, что у 2-х видов есть общий предок, у кот. произошла вставка;3) появл. вставки SINE необратимо; механизмы утраты SINE неизв-ны.4) SINE указывают на порядок возникнов-я видов в истории.Пример:по данным маркерам удалось устан-ть, что китообразные наиболее близки в происхожд. к гиппопотамам. Киты вкупе с гиппопотамовыми сост-т сестрин-ю группу со жвачными.
34. Гены, кодирующие рРНК, как основной молекулярный маркер для оценки филогенетической дистанции между организмами. ПЕРЕВОД С АНГЛ!!!!.
1. Гены, кодирующие рРНК (рДНК).
5’ (*) (*) 3’
NTS
|
ETS
|
185
|
ITS
|
5.8S
|
ITS2
|
285
|
NTS
|
** **
Кластер генов р РНК
NTS-нетранскрибируемый спейсер, ETS-внешний транскрибируемый спейсер, ITS- внутренний транскрибируемый спейсер. Спейсеры-интроны (подобие). Другие-экзоны. Такая структура свойственна эукариотам.
*-для генов дальнеродственных таксонов,
**-для близкородственных таксонов
Рибосомная ДНК (рДНК) представляет собой последов-ть ДНК, кот-ая кодируется рибосомальной РНК. рДНК эукариот состоит из тандемного повтора участка одного сегмента, оперона содержащий NTS, ETS, 18S,ITS1, 5.8S, ITS2 и 28S участки. рДНК другого гена, кодирующие 5S рРНК, располож-ые в геноме в большинстве эукариот. Последоват-сть однородно повторяющихся единиц. В большом массиве рДНК, полиморфизмы между рДНК повторя-хся звеньев явл-ся очень низкими, что указы-ет рДНК что тандемные массивы разви-ся через согласованные эволюции. Однако, механизм согласованной эволюции несовершенен, так что полиморфизм между повторами в пределах одного может вида произойти на значительном уровне и может обмануть филогенет-ие анализы для близкородс-ых органи-ов. 5S тандемный повтор последо-ти нескольких видов дрозофил были сравнены друг с другом; результатом показало, что вставки и делеции часто происходили между видами и часто в окружении консервативных последов-тей. Они могут происходить путем смещения, вновь синтезированной нити во время репликации ДНК или путем генной конверсии. Последоват-ть расхождение уточнит филогенез Транскрипции участка рДНК имеют низкую скорость полиморфизма среди видов, это позволяет сравнинить межвидовые отношения с помощью филогении лишь несколько образцов. Кодированые ITS участки рДНК высоко консервативны среди видов, но ее ITS участки явл-ся непостояными в связи со вставками, делециями и точечными мутациями. Между отдале-ми видами, как человека и лягушки сравнения последоват-тей ITS участков не подходит. Консервативные последоват-сти при кодировании области рДНК позволяют сравненить отдаленные виды, например между дрожжами и человеком. Челов-е 5.8S рРНК н а 75% идентично с дрожжами 5.8S рРНК. В таких случаях, для видов-двойников, проводят сравнение сегмента рДНК включ-ие ITS участки среди видов и филогене-ий анализ будет удовлетвор-ым. Различные кодирующие участки из rDNA повторов, как правило, показывают различные эволюцио-ые тэмпы. В результате, эта ДНК может показать филоген-ю инфор-ю о видах, принад-их к широким система-им уровням. Субъединицы рибосомы: прокари-ая и эукарио-ая рибосама может быть разбита на две субъединицы (Индекс S в представляет ед. Сведберга(скорость оседания).Прокариоты. У прокариот небольшие 30S рибосомальные субъединицы содержат 16S рРНК. Крупные 50S рибосомальные субъединицы содержат два вида рРНК (системы 5S рРНК 23S и rRNAs).Бактер-ые 16S, 23S, и 5S рРНК, и как правило, организованы в сотрудн-тве оперона транскрипции. Эукариоты. как правило эукариоты имеют много копий генов рРНК в организованных тандемных повторых; Из-за их особой стру-ры и поведения в транскрипции, кластеры генов рРНК обычно называют "рибосомной ДНК". РРНК 18S в большинстве эукариот находятся в малой рибосомной субъединице (SSU), и большой субъединице (LSU) и содержит три вида рРНК (системы 5S, 5.8S и 28S рРНК).
35. Реконструкция филогении: метод генетических дистанций.
Филогения-это описание биолог-х отношений, обычно изображаемое в виде дерева. Цель филоген-х исследований: выявление взаимосвязи между видами, популяциями, индивидами или генами. Для сопоставления необх найти гены, кот. разошлись на подходящее расстояние. Гены, которые остаются почти неизменными среди интересующих видов, не дают никакого различия в степени сходства. А гены, которые разошлись слишком сильно, не могут быть выравнены. Сущ-т 3 метода построения филогении на основании молекулярных данных::
Методы использующие оценку генетических дистанций
Методы парсимании
Вероятностные методы (м. макисм-го правдоподобия и м. Байеса)
М-ды генетических дистанций: Данная группа методов базируется на данных о генетических дистанциях. Общий принцип заключается в попарном сравнении объектов и построении матрицы дистанций, которая затем используется для построения филогенетического дерева.для дистанции прим-ся величина Р (P-distance). Это вел-на, кот описывает нуклеотидную порцию, по которой различаются виды.
Его схема: матрица данных (выравнивания) → матрица величин Р → матрица эволюционных дистанций →построение филоген-го дерева.
Собственно м. генетических дистанций поздразд-ся на:
1)М-д UPGMA или метод кластерного ан-за Метод попарного внутригруппового невзвешенного среднего (Unweighted Pair Group Method with Arithmetic Mean, UPGMA) наиб простой, широко распростр-й и имеющ высокую скорость работы, в рез-те применения кот. строится единств-е дерево с корнем (представляет соб. фенограмму). Пары послед-ей, между которыми эволюц-ые дистанции миним-ны, группир-ся и оказ-ся на соседних ветвях дерева. Здесь предпол-ся, что эволюц-е изменения происходят с равной частотой во всех сравниваемых последов-х, то длины ветвей равны половине генетического расстояния между двумя кластерами. Напр, расстояние между 1 и 2 таксономd12=5, тогда длина ветви с момента их разделения от общего предка равна d12 / 2=5 / 2=2,5. Недостатками метода: предпол-е о равномерности скорости эволюции во всех ветвях. Неравномерная скорость мутаций в сравнив-х послед-тях, приводит к неправильн. виду создаваемого дерева.
|
|
|
 Скачать 1.58 Mb.
Скачать 1.58 Mb.