1 вопр. Иммунный статус человека
 Скачать 109.99 Kb. Скачать 109.99 Kb.
|

Атаксия-телеангиэктазия Луи-БарАтаксия-телеангиэктазия Луи-Бар — наследственное заболевание, при котором, как правило, тяжёлого иммунодефицита не возникает, поэтому больные в среднем живут до 30—40 лет. Наиболее постоянный признак — низкий уровень или отсутствие IgA — встречается примерно у 70 % больных. Заболевание описано в 1941 г. Помимо иммунодефицита развиваются следующие синдромы: Церебеллярная гипоплазия (недоразвитие ткани мозжечка) проявляется нарушением координации движений (атаксией); Недостаточность ферментов репарации ДНК в клетках различных органов, вследствие чего повышается частота соматических мутаций (нестабильность генома) и нередко возникают злокачественные опухоли. Телеангиэктазии — множественные очаги расширенных мелких сосудов конъюнктивы и кожи (обнаруживаются к концу первого года жизни).Недоразвитие половых органов вследствие гипогонадизма (дефицита половых гормонов).Раннее поседение волос. Вопр. 10 ПИД с дефектами фагоцитов. Хроническая гранулематозная болезнь. Развивается в детском возрасте генетических метоболических дефектов в фагоцитах. При этом фагоциты фагоцитируют патоген, но не могут уничтожить его. В результате в них скапливаются живые микроорганизмы. Образуются гранулемы-скопления пораженных макрофагов в очаге воспаления. Для лечения применяется антибиотикотерапия. Редко-трансплантация костоного мозга. Заболевание фатально. Синдром «ленивых лейкоцитов» Миллера — совокупность наследственных дефектов функции нейтрофильных гранулоцитов: Недостаточности миграционной активности (замедленный хемотаксис) Снижения интенсивности фагоцитоза (замедленный хемотаксис и вялый фагоцитоз являются результатом нарушения функции цитоскелета, обеспечивающего локомоцию клетки и фагоцитоз) Недостаточности бактерицидной функции, прежде всего из-за дефекта кислородного механизма. Синдром «ленивых лейкоцитов» в сочетании с врождённой недостаточностью экзокринной функции поджелудочной железы называется болезнью Шва́хмана (Шве́кмана), с полным альбинизмом — болезнью Чедьяка—Хигаси Вопр. 11 ПИД с дефектом растворимых белков сыворотки крови. Дефицит компонентов комплемента. Разные компоненты комплемента кодируются разными генами. Недостаточность первых 4 компонентов комплемента С1-С4 проявляется в болезнях иммунных комплексов- системных васкулитах и повреждениях почек, что обобщенно называют синдромом системной красной волчанки. Дефицит С3 ассоциирован с повышенной восприимчивостью к пиогенным инфекциям. Дефицит С5-С9 ассоциирован с повышенной восприимчивостью к нейсериям. Дефицит С9 клинически бессимтомен. К третьей группе состояний, связанных с первичной недостаточностью комплемента, относится наследственный ангионевротический отёк Кви́нке—О́слера, в основе которого лежит недостаточность С1-ингибитора. У отдельных больных при этом возникают аутоиммунные процессы, прежде всего красная волчанка. 12, 13 вопр. Вторичные иммунодефициты— нарушения иммунной системы, развивающиеся в позднем постнатальном периоде или у взрослых, являющиеся результатом не генетических дефектов, а действия на организм целого ряда химических, радиоактивных, медикаментозных и других веществ, а также влияния вирусных инфекций, хронических воспалительных процессов, сложных операций, травм, стресса. отсутствие связи с наследственностью и генетической обусловленностью; возникновение на фоне нормальной реактивности в связи с заболеванием, воздействием неблагоприятных физических и биологических факторов, способов или средств лечения; сохранение дефицита при лечении основного заболевания и устранении факторов, индуцирующих его; отсутствие или длительная замедленная нормализация иммунного статуса. 1) бронхиты хронические, часто повторяющиеся, с пневмониями в анамнезе, сочетающиеся с заболеваниями ЛОРорганов (гнойными си нуситами, отитами, лимфаденитами); 2) пневмонии часто повторяющиеся, бронхоплевропневмонии; 3) бронхоэктатическая болезнь; 4) бактериальные инфекции кожи и подкожной клетчатки (пиодер мия, фурункулез, абсцессы, флегмоны, септические гранулемы, реци дивирующий парапроктит у взрослых); 5) грибковые поражение кожи и слизистых оболочек, кандидоз, паразитарные заболевания; 6) афтозные стоматиты в сочетании с повышенной заболеваемо стью ОРВИ; 7) рецидивирующая герпесвирусная инфекция различной локализации; 8) гастроэнтеропатия с хронической диареей неясной этиологии, дисбактериозом; 9) лимфаденопатия, повторные лимфадениты; 10) длительный субфебрилитет, лихорадка неясной этиологии; 11) генерализованные инфекции (сепсис, гнойные менингиты). Классификация вторичных ИД По форме: Наиболее ярким примером приобретенной формы ВИН является син дром приобретенного иммунодефицита человека (СПИД), развивающий ся в результате поражения лимфоидной ткани человека вирусом. Индуцированная форма ВИН возникает в результате воздействия конкретных причинных факторов: рентгеновского излучения, цито статической терапии, применения кортикостероидов Спонтанная форма ВИН характеризуется отсутствием видимой причи ны, вызвавшей нарушение иммунной реактивности. По этиологии: Стрессорные (вследствие психоэмоционального или физического стресса) •Экологические (влияние вредных факторов внешней среды) •Возрастные •При патологии I. ПО ПАТОГЕНЕЗУ: 1 нарушения преимущественно Т-клеточного звена иммунитета; 2 Нарушения преимущественно гуморального (В-звена)иммунитета; 3Комбинированные дефекты. Нарушения в системе комплемента; 5 Нарушения в системе фагоцитоза II. ПО ТЕЧЕНИЮ: •острый •хронический III. ПО РАСПРОСТРАНЕННОСТИ: • “местный” иммунодефицит; •системный иммунодефицит. IY. ПО ТЯЖЕСТИ: ЛЕГКИЙ, СРЕДНЕТЯЖЕЛЫЙ, ТЯЖЕЛЫЙ: Причины вторичных ИД Наиболее распространенными причинами, вызывающими иммунодефицит являются отравления, долгий прием определенных лекарственных средств, СВЧ и ионизирующее излучение, переутомление, хронические стрессы и загрязнение внешней среды. • Вирусные и бактериальные инфекционные заболевания, а также паразитарные инвазии • Злокачественные опухоли (новообразования), которые нарушают все системы организма. Более явное снижение иммунитета проявляется в замещении костного мозга опухолевыми метастазами и при злокачественных болезнях крови (лейкемии). На фоне лейкемии число иммунных клеток в крови увеличивается во много раз. Однако они не могут обеспечить защитную функцию, так как являются нефункциональными. • Аутоиммунные болезни, которые формируются в результате сбоя работы иммунной системы. • Нарушение питания, истощение организма, которые приводят к снижению иммунитета. Белково-калорическая недостаточность оказывает выраженное негативное влияние на лимфоидную ткань и клеточный иммунитет. •Иммуномодулирующие лекарственные препараты могут сильно подавлять иммунные функции.Стероиды влияют на миграцию клеток, индуцируют лейкоцитопению и ингибируют синтез цитокинов. • Утрата факторов иммунной защиты, которая наблюдается при ожогах, болезнях почек и сильных кровопотерь. • Эндокринные болезни, которые приводят к понижению функции иммунной системы в результате сбоя обмена веществ. При сахарном диабете увеличивается частота инфекционных болезней, что связано с угнетением работы иммунной системы и высоким содержанием глюкозы в составе крови, что способствует размножению бактерий. • Серьезные операции и травмы, которые протекают с понижением работоспособности иммунной системы. • Прием наркотических и лекарственных препаратов, которые оказывают интенсивное иммунодепрессивное воздействие. • Снижение иммунитета у пожилых людей, детей и беременных, что связано с физиологическими или возрастными особенностями организма. • Вич-инфекция 15, 16 вопр. ВИЧ-инфекция Возбудителем СПИДа является вирус иммуноде-фицита человека (ВИЧ) - РНК-содержащий ретровирус, который связывается с CD4. В результате происходит значительное снижение числа Т-клеток CD4+ (Т-хелперов), что ведет к резкому нарушению клеточного иммунитета и к гибели от оппортунистических инфекций. ИЧ относится к семейству ретровирусов, его геном состоит из двух идентичных копий одноцепочечной РНК. Вирус имеет специальный фермент - обратную транскриптазу (RT - Reverse Transcriptase),также именуемую ревертазой. Ревертаза после проникновения вируса в клетку-мишень синтезирует цепь ДНК на матрице вирусной РНК, т.е. этот фермент действует как РНК-зависимая ДНК-полимераза. • Другой вирусный фермент - интеграза - катализирует ковалентную интеграцию вирусной ДНК в ДНК генома человека, причём сразу в несколько разных участков. Интегрированная ДНК вируса называется провирусом. • Клинически латентный период сильно различается по продолжительности у разных людей, что зависит от дозы полученного при заражении вируса, генотипа человека (HLA), его исходного состояния здоровья, наличия других вирусных и бактериальных инфекционных заболеваний, образа жизни., острая лихорадочная фаза, бессимптомная, СПИД Выделяют несколько групп пациентов по продолжительности клинически латентного периода ВИЧ-инфекции: ◊ у 10% заболевание развивается до стадии СПИД в первые 2-3 года от момента заражения;◊ у 80-85% людей заболевание развивается до стадии СПИД в течение 10 лет;◊ у 5-10% инфицированных людей клинические симптомы отсутствуют в течение 7-10 лет, при этом сохраняется стабильный уровень CD4+ T-лимфоцитов в крови. Диагностика• РНК и ДНК вируса (методом ПЦР);• противовирусных антител (методами иммуноферментного анализа, иммунохроматографии и иммуноблоттинга);• белка p24 (с помощью «ловушечного» иммуноферментного анализа);• живого реплицирующегося вируса (высев в культуру клеток in vitro). Лечение •Нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы. абакавир; абакавир + ламивудин + зидовудин; диданозин;•Ненуклеозидные ингибиторы обратной транскриптазы. Невирапин; эфавиренз; делавирдин.•Ингибиторы вирусной протеазы: ампренавир• Ингибиторы слияния вириона с клеткой-мишенью: энфувиртид.• Ингибиторы интегразы: ралтегравир.• Ингибиторы созревания. • Препараты, подавляющие синтез ДНК: В настоящее время с помощью определённой комбинированной противовирусной химиотерапии можно достичь быстрого снижения репликации ВИЧ в организме и соответственно уменьшения вплоть до неопределимых анализами количеств вирусных РНК в крови на период до года. Такая химиотерапия получила название высокоактивной антиретровирусной медикаментозной терапии - HAART (Highly Active AntiRetroviral Therapy).HAART включает 1 или более ингибитор вирусной протеазы и 2 или более ненуклеозидных ингибитора обратной транскриптазы вируса. При развившейся ВИЧ-инфекции иммунотерапия не показана. Дело в том, что именно гиперстимуляция иммунной системы самой вирусной инфекцией и приводит к повреждению иммунной системы 17 вопр. Аутоиммунные болезни — это болезни, в патогенезе которых лимфоциты распознают нативные молекулы мембран собственных клеток или межклеточного вещества и инициируют иммунное воспаление. Причины АБ На развитие аутоиммунных заболеваний влияют: 1.наследственная предрасположенность (наследование определенных генов HLA, генов иммуноглобулинов и антигенраспознающего рецептора T-лимфоцитов) 2. действие факторов окружающей среды (ультрафиолетовое излучение при СКВ, бактериальная инфекция при реактивных артритах) 3. нарушения иммунитета. Аутоантигенами могут стать любые ткани, клетки и компоненты плазмы, иммуноглобулины. Так, ревматоидный фактор, например, — это аутоантитела к IgG. К аутоиммунным заболеваниям не относятся: 1. Патологические процессы, при которых имеется повреждение тканей иммунными механизмами. Но это будут не аутоиммунные процессы, поскольку направлены против микробных антигенов и разрушение тканей наступает потому, что микробные продукты оказались тесно связанными с нашими тканями. 2. Денатурация собственных молекул организма химическими веществами. иммунная система борется с внешними повреждениями на поверхности собственных тканей. В норме у каждого здорового организма в периферических лимфоидных тканях есть и Т-, и В-лимфоциты с антигенраспознающими рецепторами для «своего», т.е. аутоиммунные болезни не является результатом возникновения аномальных лимфоцитов: они всегда присутствуют. Иммунный Т-лимфоцит отличается от неиммунного Т-лимфоцита: 1) для активации эффекторной функции иммунному лимфоциту достаточно только сигнала с его рецептора 2) молекулы адгезии иммунного лимфоцита позволяют ему мигрировать в любые периферические ткани, а неиммунный лимфоцит рециркулирует строго между «своими» зонами в периферических лимфоидных органах. Поэтому инфекции способны инициировать аутоиммунные процессы по следующим механизмам: • 1.Антигенная мимикрия патогенов Размножаясь внутри клеток организма,вирусы время от времени включают в состав своего генома какие-то из генов этого организма. • 2. Микробные суперантигены вызывают поликлональную активацию. Некоторые клоны лимфоцитов с реактивностью к своим антигенам могут войти в режим эффекторного иммунного ответа. • 3. Деструкция тканей патогеном (цитопатогенное действие вирусов, бактерий и др.) приводит к попаданию тканевых антигенов в активированные дендритные клетки, которые транспортируют все антигены в периферические лимфоидные органы, где есть условия для индукции продуктивного иммунного ответа • 4. Индуцированное патогеном локальное доиммунное воспаление сопровождается выработкой провоспалительных цитокинов, которые способны индуцировать экспрессию на клетках тканей молекулы МНС со своими пептидами, что потенциально создает условия для инициации иммунного ответа на свои антигены. • 5. Два рецептора на одном Т-лимфоците. Есть вероятность, что один из них может иметь специфичность к патогену, а второй — к аутоантигену. клона иммунных лимфоцитов, которые будут работать в качестве эффекторов против обоих «своих» антигенов — чужого и своего. Ассоциация аутоиммунных болезней с определенными антигенами МНС может быть понята именно с учетом «инфекционного» компонента патогенеза, так как именно МНС представляют антигены для распознавания Т-лимфоцитам: как представят, такой иммунный ответ и будет. При некоторых заболеваниях этиология иммунного воспаления связана не с конкретными антигенами, а с нарушениями в нормальных механизмах апоптоза лимфоцитов. Например, при ревматоидных артритах, иммунное воспаление суставов вызвано тем, что зрелые иммунные Т-лимфоциты в синовиальных полостях своевременно не погибают апоптозом, а продолжают продуцировать провоспалительные цитокины, потому что сами получают патологический сигнал на выживание от измененных фибробластов стромы синовиальных хрящей Классификация АБ 1. Органоспецифические болезни, которые вызываются аутоантителами и сенсибилизированными лимфоцитами против одного или группы аутоантигенов конкретного органаК ним относятся тиреоидит Хосимото, миастения гравис, 2. При неорганоспецифических системную красную волчанку, дискоидную эритематозную волчанку, ревматоидный артрит, дерматомиозит (склеродермия). 3. Смешанные болезни включают оба перечисленных механизма. сахарный диабет 1 типа 18 вопр. Теории развития аутоиммунных болезней Теория расстройства иммунологической регуляции. Предполагает, что снизились количество или функции каких-то клеток, отвечающих за подавление иммунного ответа - либо Т-супрессоров, либо Т-хелперов, либо нарушена продукция цитокинов этими клетками. Теория "запретных" клонов. В тимусе и костном мозге по каким-то причинам не происходит полная элиминация аутореактивных Т- и В-лимфоцитов, что приводит к агрессии против собственных тканей. Теория секвестрированных антигенов. Ряд тканей организма защищены гистогематическими барьерами (половые железы, ткани глаза, мозга, щитовидной железы и др.). Поэтому при формировании иммунной системы антигены таких тканей не контактируют с лимфоцитами и не происходит элиминации соответствующих клонов. При нарушении гистогематического барьера и попадании антигенов в кровоток иммунные клетки воспринимают их как чужих и начинают иммунную реакцию. Теория нарушения идиотип-антиидиотипических взаимодействий (теория Эрне). Предполагает, что регуляция иммунной системы осуществляется в основном антииммуноглобулинами. Соответственно нарушение идиотип-антиидиотипических взаимодействий приводит к аутоиммунным заболеваниям. Теория поликлональной активации В-лимфоцитов. Многие вещества (например, некоторые антибиотики, вирусы, ферменты бактерий и т.п.) могут индуцировать активацию В-лимфоцитов, которая приводит к их пролиферации и продукции антител. Если активированы В-лимфоциты, продуцирующие аутоантитела, возникают аутоиммунные заболевания. Теория развития аутоиммунитета под влиянием суперантигенов. Суперантиген активирует лимфоциты независимо от их антигенной специфичности, что приводит к активации также и аутореактивных лимфоцитов. Подобные антигены производятся рядом бактерий. Теория генетической предрасположенности. Подозреваются шесть генов, расположенных на разных хромосомах. женщины подвержены аутоимунным заболеваниям чаще, чем Вопр. 19 Признаки аутоиммунных заболеваний, сформулированы еще Л. Витебски (1961). присутствие аутоантител или Т-киллеров, направленных против антигена, ассоциированного с данным заболеванием; возможность переноса аутоиммунитета с помощью сыворотки, которая содержит антитела или Т-киллеры; идентификация аутоантигена, против которого направлена иммунная реакция; возможность создания экспериментальной модели заболевания с помощью введения аутоантигена вопр. 20 Общие принципы иммуно-лабораторной диагностики аутоиммунных заболеваний: 1. Поиск специфических аутоантител 2. Определение специфической клеточной сенсибилизации (с помощью реакции бласттрансформации и теста ингибиции миграции лейкоцитов в присутствии соответствующего аутоантигена) 3. Повышение уровня гамма-глобулина и/или IgG 4. Изменение количества Т-хелперов и Т-супрессоров, приводящее к повышению иммунорегуляторного индекса 5. Снижение уровня СЗ и С4 компонентов комплемента 6. Отложения иммунных комплексов в пораженных тканях (IgG, IgM, СЗ, С4 и фибрин) 7. Лимфоидно-клеточная инфильтрация пораженных тканей 8. Определение HLA-фенотипа. Вопр. 21 АБ эндокринных желез Аутоиммунный гипертироидизм [болезнь Грейвса (Grawes)].. Является наиболее частой причиной гипертироидизма. В 90 % случаев удается обнаружить антитела к рецептору для тиростимулирующего гормона гипофиза. Эти антитела вместо гормона стимулируют функциональную активность клеток щитовидной железы, что и приводит к развитию симптомокомплекса гипертироидизма. Лечение. Иммунодепрессанты в тяжелых случаях экзофтальма. Обычно - антитироидные блокаторы. Хирургическое удаление части щитовидной железы. Инсулинзависимый диабет (ИЗД) (1 типа). ИЗД развивается в результате селективного разрушения β -клеток панкреатических островков (островков Лангерганса) CD8+ ЦТЛ и CD4+ Thl-опосредованным иммунным воспалением. Этиология болезни предположительна. Прослеживаются клинические ассоциации с рядом вирусных инфекций (краснуха, вирус коксаки , реовирусы и др.), а также с интоксикацией некоторыми химическими соединениями Клинически болезнь манифестирует «остро» симптомами иолиурии, полидипсии и быстрого похудания. Процесс разрушения β-клеток протекает в течение нескольких лет до этого и при жизни не диагностируется в связи с компенсированностью клинической картины. В преклинической стадии диагностическое значение имеет определение в сыворотке крови антител к клеточным антигенам β -клеток, антиинсулиновых антител, антител к глютаматдекарбоксилазе (анти-GAD) и антител к тирозинфосфатазе ΙΑ-2, а также тесты на толерантность к глюкозе (внутривенный и пероральный). Лечение . Заместительный инсулин. Аутоиммунный зоб В начальный период болезни Хасимото у 75% пациентов функциональный статус щитовидной железы в пределах нормы, у 20% - гипотиреоидизм, у 5% - гипертиреоидизм. Первые признаки заболевания обнаруживают, как правило, в подростковом возрасте, но клинически значимое состояние развивается в большинстве (90%) случаев после 45 лет. По мере прогрессирования заболевания у 50% пациентов развивается гипотиреоидизм. Более чем у 95% больных выявляют антитела к тиреоидной пероксидазе (микросомный антиген), иногда к тиреоглобулину. Лечение. Заместительная терапия тиреоидными гормонами (тироксин и трийодтиронин) под контролем уровня этих гормонов в сыворотке крови. Недостаточность надпочечников Болезнь Аддисона выявляют с частотой 6:100 000 населения, у женщин в 2 раза чаще. Поражается кора, но не мозговое вещество надпочечников. У 80% больных обнаруживают антитела к 17а-гидроксилазе. Лечение. Заместительная терапия гормонами коры надпочечников. Болезнь Кушинга Болезнь Кушинга - микронодулярная адренокортикальная гиперплазия. Вызвана антителами к рецептору адренокортикотропного гормона, которые стимулируют надпочечники вместо этого гормона. Как правило, у 50% больных выявляют множественные эндокринопатии и антитела к другим тканевым антигенам: у 20% - патологию щитовидной железы, у 15% - сахарный диабет, у 8% - патологию яичников, у 4% - гипопаратиреоидизм. Лечение симптоматическое. Вопр. 22 АБ ЖКТ Гастрит и пернициозная анемия. Встречаются с частотой 0,1 % в западных странах. Характеризуются атрофией слизистой оболочки желудка с потерей париетальных и chief-клеток. У 90 % больных обнаруживают антитела к мембранному белку париетальных клеток и у 70 % — к внутреннему фактору для витамина В12. Слизистая оболочка инфильтрирована лимфоцитами (CD4+, CD8+, В). Лечение симптоматическое. Воспалительные заболевания кишечника (Inflammatory bowel disease). Этим «сборным» термином обозначают идиопатический язвенный колит и нечетко отличающуюся от него болезнь Крона (Crohn). Гистологическая картина: трансмуральное воспаление с лимфоидными агрегатами и подозревают инфекцию, особенно в случае болезни Крона. характерна гипертрофия нервов и подслизистого мезентериального сплетения. При болезни Крона симптомы зависят от конкретной локализации и размеров воспаления: лихорадка, боли в животе, диарея, потеря массы тела. Возможны симптомы обструкции, фистулы, абсцессы. В крови больных определяют антитела к пищевым антигенам и кишечным бактериям-комменсалам, но это рассматривают как результат патологически повышенной проницаемости воспаленного кишечника. Определяют также антитела, связывающиеся с поверхностными структурами эпителиальных клеток толстой кишки. Эти антитела перекрестно реагируют с микробными продуктами E.coli. Лечение . Сульфасалазин, аминосалицилаты, в тяжелых случаях кортикостероиды. В 80 % случаев болезни Крона возникает необходимость в хирургическом вмешательстве. Вопр. 23 АБ НС Рассеянный склероз. При этом заболевании происходит диссеминированная демиелинизация аксонов мозга. Бляшки демиелинизации бывают размером от 1 мм до нескольких сантиметров. Олигодендроциты разрушаются, астроциты избыточно пролиферируют, в области бляшек развивается ацеллюлярный фиброз. В периваскулярных областях отмечается лимфоцитарная инфильтрация. Аутоантиген — МВР — главный основный протеин миелина. Иммунопатогенез состоит в повреждении миелина по механизму ГЗТ. Клиническая картина. Характерны симптомы неврита зрительного нерва, головокружения, нарушения координации, спинальные синдромы у 30 % больных. При прогрессировании — спутанное сознание, депрессия, деменция. Диагноз ставят только по клинической картине. Адекватных лабораторных методов дифференциальной диагностики нет. Лечение. Адекватного лечения нет. У некоторых пациентов отмечают временный эффект от больших доз метилпреднизолона. Иммунодепрессанты неэффективны. Прогноз. Через 15 лет от момента манифестации 10 % пациентов не могут обходиться без инвалидного кресла, 50 % вынуждены пользоваться палкой или посторонней помощью при ходьбе. Миастения гравис (Myasthenia gravis). Заболевание описано в 1672 г. Th. Willis. Получило название myasthenia gravis (слабость «до гробовой доски») в 1895 г. (F. Jolly). Тимэктомия в лечебных целях впервые была сделана в 1911 г. Заболевание встречается в Европе с частотой 2—4 на 100 000 населения, чаще у женщин. Пик манифестации — в возрасте 20—30 лет. Этиология неясна. Патогенез известен: обусловлен блокирующими антителами к рецептору для ацетилхолина, обеспечивающему передачу нервного импульса с двигательных нервов на поперечнополосатые мышцы. У большинства пациентов с myasthenia gravis есть аномалии в тимусе: в 70 % случаев представляют собой лимфоидную фолликулярную гиперплазию. В 10 % случаев обнаруживают доброкачественную тимому. При этом опухолевые клетки тимуса имеют признаки подобия клеткам поперечнополосатых мышц. Степень выраженности клинических симптомов: от средней степени до тяжелой мышечной дебильности и дыхательной недостаточности при вовлечении в процесс мышц, участвующих в респираторных движениях. Лечение. Ингибиторы ацетилхолинэстеразы, тимэктомия, стероиды, иммунодепрессанты. Прогноз. Еще 30 лет назад от myasthenia gravis умирал каждый 4-й больной. Риск летального исхода максимален в течение 1-го года после постановки диагноза. Если человек переживает этот срок без признаков быстрой прогрессии, то прогноз благоприятен. Если в первые 2 года после манифестации произведена тимэктомия, то у некоторых пациентов достигается перманентная ремиссия. Если в течение 7 лет заболевание при любом способе лечения не выходит в «режим» быстрой прогрессии, то риск тяжелого релапса невелик. Вопр. 24 АБ крови Аутоиммунная гемолитическая анемия. Различают две формы: тепловую и холодовую. При тепловой гемолитической анемии (протекающей при нормальной температуре внутренней среды организма, 36,8—37°С) эритроциты человека аномально покрыты антителами преимущественно класса G и компонентами комплемента СЗ и С4. В таком виде эритроциты подвергаются повышенной деструкции макрофагами печени и селезенки. Этиология неизвестна. Лечение. В первую очередь лечат основную болезнь. При тяжелой анемии показаны гемотрансфузии; иногда улучшение состояния достигают применением глюкокортикоидов, иммуносупрессивных препаратов, спленэктомии. Холодовую гемолитическую анемию вызывают антитела класса М, обычно направленные против антигенов эритроцитов. У пожилых пациентов холодовая гемолитическая анемия чаще всего является осложнением лимфопролиферативных процессов и имеет хроническое длительное течение. Антиэритроцитарные антитела присоединяются к эритроцитам только в периферических сосудах, где температура крови ниже 32 °С, затем в глубоких сосудах комплекс эритроцит — антитела фиксирует комплемент и развивается внутрисосудистый гемолиз. Гемолиз происходит эпизодами в связи с переохлаждением и ознобами, может сопровождаться желтухой и гемоглобинурией. Лечение. Избегание переохлаждений. Лекарственный гемолиз Отдельные лекарственные средства у некоторых людей могут сорбироваться на поверхности эритроцитов или образовывать комплексы с белками крови, в том числе и с иммуноглобулинами. Гемолитическая болезнь новорождённых Гемолитическая болезнь новорождённых - экстраваскулярное разрушение эритроцитов плода антиэритроцитарными антителами матери, проникшими через плаценту (IgG). Как правило, гораздо сильнее проявляется так называемый резус-конфликт в случае Rh-отрицательной матери и Rh-положительного плода. для профилактики резус-конфликта при текущей беременности женщине вводят aнти-Rh(D)-cывopoткy - что через FcyRIIB на B-лимфоцитах подавляет образование антител B-лимфоцитами именно клона aнти-Rh(D). Вопр. 25 Аут.б. Первичные системные васкулиты Первичными системными васкулитами называют заболевания, при которых развиваются воспаление и некроз стенок сосудов, а патологические процессы в окружающих тканях вторичны по отношению к поражению сосудов. В связи с неясностью этиологии васкулиты до настоящего времени классифицируют по морфологическим признакам, по калибру поражаемых сосудов и наличию или отсутствию гранулем вокруг пораженных участков сосудов . Этиология ни одного из этих васкулитов неизвестна. Общие признаки васкулитов: цикличность чередования обострений и ремиссий, сезонность обострений (в холодное время года) и общность неспецифических симптомов -общая слабость, лихорадка, потеря массы тела, ночные поты, повышение СОЭ и содержания в сыворотке С-реактивного протеина — СРП). Лечение . При системных васкулитах оно одинаково, иммунодепрессанты и высокие дозы стероидов; поддерживающую терапию — стероиды плюс симптоматические средства в зависимости от локализации процесса и присоединившихся патологий. В периоды ремиссий — отдых от медикаментов. Поскольку при всех васкулитах повреждаются почки, в тяжелых случаях единственным методом спасения жизни становится трансплантация донорской почки. Гранулематоз Вегенера (Wegener). Заболевание встречается в любом возрасте, одинаково распределено по полу. У 85 % больных гранулематозом Вегенера обнаруживают антитела к сериновой протеиназе-3 (РгЗ) нейтрофилов. Клиническая картина. Некротизизующий васкулит мелких сосудов развивается в верхних и нижних дыхательных путях, поражает почки, может затронуть другие органы. У некоторых пациентов заболевание развивается бессимптомно в течение многих месяцев и даже лет.Верхние дыхательные пути: выделения из носа с примесью крови, боли в параназальном синусе, изъязвление слизистой оболочки носа, перфорация перегородки и деформация носа. Возможно развитие хронического гнойного среднего отита в связи с блокадой слуховой трубы, боли по ветвям VIII нерва. Возможны осиплость голоса и стеноз трахеи. В синусах могут образовываться фистулы с бактериальными суперинфекциями. Нижние дыхательные пути: гранулемы в паренхиме легких вызывают симптомы непродуктивного кашля, диспноэ, плеврита, кровохарканья. Возможны окклюзии бронхов и сегментарные коллапсы легких. Васкулит в почках начинает проявляться как клинически бессимптомная протеинурия и гематурия. При биопсии почечной ткани наблюдают картину гранулематозного нефрита. Около 50 % больных гранулематозом Вегенера имеют поражение глаз вплоть до потери зрения. Поражения других органов развиваются позже. Диагноз ставят по клиническим симптомам и данным биопсии. Лабораторным подтверждением является обнаружение антител ANCA. тромбоцитоз, повышение СОЭ , у 80 % больных увеличено содержание общих иммуноглобулинов в сыворотке крови, у 50 % определяется ревматоидный фактор. Вопр. 26 Аллергены - это антигены, вызывающие специфически повышенную чувствительность организма - аллергию. Аллергены делятся на экзогенные, попадающие в организм из внешней среды, и эндогенные, имеющиеся или образующиеся в самом организме. Экзогенные аллергены по происхождению делятся на инфекционные и неинфекционные. 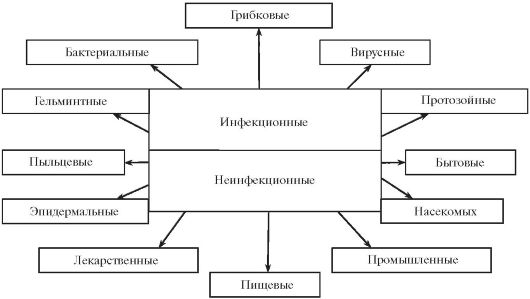 Аллергены отличаются некими особыми физическими и химическими свойствами, а также особенностями поступления в организм, т.е. аллергены — как бы подмножество во множестве антигенов вообще. Если аллергены — белки, то чаще всего это ферменты протеазы. Аллергены имеют относительно невысокую молекулярную массу, способны сорбироваться или агрегироваться в мелкие частицы и в таком виде проникать в слизистые секреты и покровные ткани , без видимого травмирования покровных тканей. При этом аллергены хорошо растворимы и легко элюируются в жидкие среды организма. Если аллергены — не белки , то их отличает способность вступать в химические соединения с собственными белками организма. (бром, йод, хром, никель). Эти вещества называют гаптенами. Аллерген проявляет свое действие в крайне малых дозах, например патогенно значимая суммарная доза аллергенов амброзии может составлять всего 1 мкг за год. Вопр. 27 Иммунологическая, патохимическая, патофизиологическая Вопр. 28 Эпидемиология аллергических болезней Самое интересное, что природное место основной массы IgE не в крови (в крови остаётся менее 1% синтезированного IgE), более 99 % IgE организм декретирует через эпителий ЖКТ в просвет кишки. Просвет кишки - наиболее известное место большинства известных гельминтных инвазий Эпидемиологические исследования показали, что у людей прослеживается сильная обратная корреляция между гельминтозами и аллергическими болезнями.—- чем меньше гельминтозов, тем больше аллергических болезней Естественно приобретаемые гельминтозы в детстве обеспечивают такое развитие пропорций в субпопуляциях лимфоцитов, которое на всю оставшуюся жизнь предохраняет; организм, например, от аллергий. Причины - снижение уровня или полная ликвидация эпи заболеваний уменьшило контакт с сильными аг, введение вакцин и сывороток способствует повышению сенсибилизации, возросло число новых хим веществ, изменение условий жизни и питания. классификация гиперчувствит 1, 2, 3, 4 типа (ГЗТ) Вопр. 29 Истинная аллергия, ниперчувствительность 1 типа Вопр. 30 Бронхиальная астма При этом заболевании наблюдается хроническое воспаление слизистой оболочки дыхательных путей с синдромом гиперреактивности бронхов (ГРБ). Определяющие клинические симптомы: кашель, «свистящее» дыхание, затрудненный выдох, поверхностное дыхание, возможны диспноэ, потеря голоса, цианоз и т.д. Для всех форм заболевания характерно повышение неспецифической реактивности бронхов (НРБ), выражающееся в том, что приступ обструкции дыхательных путей может возникнуть в ответ на разные раздражители типа холодного воздуха, неорганической пыли, табачного дыма, запахов парфюмерии, красок и т.п. Механизм развития хронического воспалительного процесса в дыхательных путях при АБА заключается в патологическом сдвиге дифференцировки аллергенспецифических Т4-лимфоцитов в сторону Th2 в лимфоидной ткани слизистой дыхательных путей. При попадании специфического аллергена в дыхательные пути у больного развивается либо только однофазная ранняя реакция бронхоспазма, которая заметна уже через 5—10 мин (с пиком через 15—20 мин) от момента попадания аллергена в дыхательные пути (ОРФ — ответ ранней фазы), либо, кроме ОРФ, через 3—9 ч (с пиком в среднем на 5-м часу) развивается еще ответ поздней фазы (ОПФ). ОРФ разрешается через 1—2 ч, ОПФ продолжается от нескольких часов до нескольких суток. В отличие от ОРФ ОПФ объясняется не спазмом крупных дыхательных путей, а закупоркой множества мелких дыхательных путей. Наличие ОПФ свидетельствует о более тяжелой клинической форме бронхиальной астмы, чем в случаях, когда есть только ОРФ. Механизмы развития ОРФ и ОПФ зависят от аллергенспецифических IgE и тучных клеток. Только ОРФ — результат действия медиаторов гранул тучных клеток, выбрасываемых немедленно после связывания аллергена с IgE на FcЕRI, в первую очередь гистамина. Поэтому симптоматика ОРФ может быть купирована антигистаминными препаратами. ОПФ — результат действия медиаторов тех же активированных тучных клеток, но других медиаторов, для синтеза и секреции которых требуется несколько часов, — метаболитов арахидоновой кислоты (лейкотриены, простагландины, PAF) и цитокинов. В патогенезе бронхиальной астмы эозинофилам принадлежит особая роль как клеткам-эффекторам деструкции тканей. Диагноз аллергической бронхиальной астмы ставят по совокупности данных анамнеза, физикально выявляемым симптомам, по данным лабораторного анализа на аллергенспецифические IgE или/и кожных проб с предполагаемыми аллергенами, инструментального обследования параметров функции внешнего дыхания. Подозрение именно на аллергическую этиологию бронхиальной астмы возникает, если у пациента уже есть диагноз других проявлений атопии, в первую очередь аллергического ринита. Лечение. Уже много лет основным лечением бронхиальной астмы являются кортикостероиды. Бронходилататоры — это средства симптоматической терапии Адреналин не является средством поддерживающей терапии при бронхиальной астме, но, как при всех аллергических болезнях, он является средством спасения жизни в случае острого состояния с угрозой или явлениями системной анафилаксии Вопр. 31 Системная анафилаксия — наиболее драматическое клиническое проявление массивного высвобождения медиаторов из гранул тучных клеток и базофилов. Она характеризуется быстрым наступлением симптомов (в пределах 30 мин от момента воздействия причинного фактора) во многих органах и потенциально смертельно опасна. Собственно анафилаксией принято называть патологический процесс, инициация которого происходит через взаимодействие антиген — IgE-FcЕRI — тучная клетка/базофил. Но дегрануляция тучных клеток/базофилов может происходить под действием множества других факторов, кроме «антигена-IgE». IgE-независимые процессы принято называть анафилактоидными. Клиническая картина. Анафилаксия развивается через несколько минут после причинного воздействия, и смерть может наступить через несколько минут. Процесс может затянуться и на несколько часов и даже дней. Патологоанатомически наблюдают следующее: 1) отек верхних дыхательных путей (включая гортань), что может привести к острому удушью;2) усиленную секрецию слизи в нижних дыхательных путях, отек, застой крови, эозинофильную инфильтрацию;) отек легких, иногда геморрагии ;4) повышение проницаемости сосудов, отеки в тканях и уменьшение объема крови внутри сосудов, резкое падение кровяного давления, «побледнение» сердечной мышцы (ухудшение кровоснабжения миокарда), ишемию почек и других внутренних органов;5) застойные явления в печени, селезенке, стенке кишечника;6) отек кожи;7) спазмы гладкой мускулатуры мочевыводящих путей и кишечника. Проявления со стороны респираторной системы превалируют в 70 % случаев системной анафилаксии, со стороны кардиоваскулярной системы — в 25 % случаев. Из «причинных» аллергенов системную анафилаксию вызывают, как правило, 3 типа: яды жалящих насекомых (перепончатокрылых — пчел и ос), пищевые аллергены и лекарственные препараты. Лечение. В остром периоде системная анафилаксия требует экстренных, часто реанимационных мер. Если ведущим симптомом является бронхоспазм, то в первую очередь применяют аэрозольные формы бронходилататоров . Если симптоматика тем не менее продолжает нарастать и присоединяются сосудистые явления, то применяют кислородную маску и адреналин внутримышечно с интервалами 5—10 мин под контролем ответа организма. Внутривенное введение адреналина резервируют на случай развития тяжелого шока или респираторной асфиксии (кроме того, внутривенное введение адреналина может вызвать аритмию сердца). В остром периоде антигистаминные препараты и кортикостероиды неэффективны, и применять их не имеет смысла. Однако для профилактики поздней фазы стероиды иногда используют. Вопр. 32 Крапивница и ангиоэдема (ангионевротический отек). характеристика основные признаки причины Крапивница (уртикария) — очень распространенное аллергическое кожное заболевание. Такое название объясняется высыпанием зудящих волдырей, как при ожоге крапивой. Крапивницу узнают по характерной сыпи. На коже образуются вздутия или волдыри, бледные в центре и красные по краям. Волдыри и окружающая их кожа сильно чешутся. Заболевание поражает только поверхностные участки кожи. Сыпь при крапивнице обычно сохраняется несколько часов, редко дольше 48. Крапивница возникает внезапно под действием какого-нибудь стимула или периодически возвращается в виде повторных рецидивов в течение ряда месяев и даже лет без всякой видимой причины. Если заболевание, периодически обостряясь, длится не более месяца, его относят к острым, но если вспышки крапивницы повторяются в течение более длительного времени, то заболевание считается хроническим. |