Айтказинова Айгерим ИМА 14нед. 14ші аптаны Cенбікнісаат
 Скачать 41.38 Kb. Скачать 41.38 Kb.
|

|
БІЛІМДІ ТЕКСЕРУГЕ АРНАЛҒАН СҰРАҚТАР Cоңғы тапсыру мерзімі 14-ші аптаның Cенбікүнісағат 19:00-ге дейін! Тапсырма №8 1. Гравиметриялық талдаудағы түрлендіру коэффициенті дегеніміз не? В гравиметрическом анализе обычно находят не абсолютное количество определяемого компонента, а его содержание (масс. доля, %) в анализируемом веществе. Гравиметрический анализ включает два экспериментальных измерения: взвешивание навески и взвешивание продукта известного состава, полученного из этой навески. На основании этих данных обычно рассчитывают содержание определяемого компонента. Если А – определяемый компонент, можно записать 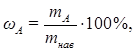 где, ωА - массовая доля (%) определяемого компонента; mнав – масса навески образца анализируемого вещества; mА – масса определяемого компонента в анализируемом образце. Например, содержание влаги (влажность) определяют, взвешивая навеску влажного образца до и после высушивания. Массу удаленной при высушивании воды рассчитывают по разности: m(H2O) = m(влаж нав) - m(сух нав); m(влаж нав) – масса влажной навески анализируемого вещества; m(сух нав)– масса этой же навески после высушивания. Содержание воды (влаги, влажность) рассчитывают по соотношению  Чаще всего масса определяемого компонента непосредственно не измеряется. В методах осаждения выделяют и взвешивают вещество, либо содержащее, либо химически связанное с определяемым компонентом. Расчеты результатов гравиметрического анализа базируются на основных стехиометрических законах химии и могут быть проведены по уравнениям реакций. Чаще всего масса определяемого компонента непосредственно не измеряется. В методах осаждения выделяют и взвешивают вещество, либо содержащее, либо химически связанное с определяемым компонентом. Расчеты результатов гравиметрического анализа базируются на основных стехиометрических законах химии и могут быть проведены по уравнениям реакций.2. Йодометрияда индикатор қызметін не атқарады? Мысал келтіріңіз. В качестве индикатора в йодометрии используется водный раствор крахмала, который образует с молекулярным йодом йодкрахмальное соединение синего цвета. При титровании восстановителей рабочим раствором йода точка эквивалентности определяется по появлению интенсивно-синего окрашивания. При титровании йода рабочим раствором тиосульфата натрия конец реакции определяется по исчезновению синей окраски от одной капли раствора тиосульфата натрия. Крахмал необходимо добавлять в самом конце титрования, когда йода в растворе становится мало и раствор приобретает соломенно-желтый цвет. Крахмал, добавленный к раствору с высокой концентрацией йода, становится черным и разрушается, что вносит ошибку в определение точки эквивалентности. Очень высокая чувствительность крахмала к йоду и резкое изменение окраски раствора в точке эквивалентности позволяют использовать рабочие растворы меньшей концентрации, чем в других методах титриметрического анализа (от 0,01000 н. до 0,05000 н.). Многие йодометрические реакции обратимы и протекают до конца только при создании следующих условий: 1) титрование необходимо проводить на холоде, так как, во-первых, йод - летучее соединение и, во-вторых, при нагревании чувствительность крахмала к йоду уменьшается; 2) рН раствора не должен превышать 9, так как в щелочной среде йод диспропорционирует: I2 + 2ОН- ↔ I- + IO- + Н2О, а IO-, являясь более сильным окислителем, чем I2, окисляет тиосульфат-ион до сульфат-иона: S2O2-3 + 2OН- + 4 IO- = 2SO2-4 + Н2О + 4I-. Большим преимуществом йодометрического метода является доступность чувствительного и обратимого к йоду индикатора. К недостаткам относится низкая устойчивость раствора йода и неполное протекание реакций йода со многими восстановителями. Однако по простоте и точности метод йодометрии считается одним из лучших. В клиническом анализе им пользуются при определении сахара в крови, в медико-гигиеническом анализе - для определения «активного» хлора в хлорной извести и свободного хлора в воде. В фармацевтическом анализе метод используется для определения концентрации свободного пода, количества йодидов и тиосульфата натрия. Таким образом, метод йодометрии может использоваться для определения как окислителей, так и восстановителей. Рассмотрим подробнее их определение. Йодометрическое определение восстановителей проводят по методу прямого или обратного титрования. Окислители определяют путем заместительного (косвенного) титрования. При определении восстановителей методом прямого титрования рабочим раствором является раствор йода. Этим методом определяют соединения мышьяка (III), сурьмы (III), олова (II), тиосульфаты, небольшие количества H2S (например, в минеральных водах), сульфидов и сульфитов. В качестве примеров реакций, протекающих при прямом титровании восстановителей йодом, можно привести следующие: SO2-3 + I2 + Н2O = SO2-4 + 2I- + 2Н+; AsO2 + I2 + 2Н2О = HAsO2-4 + 2I- + 3Н+. В тех случаях, когда прямое титрование осложнено (например, восстановитель летучий или реакция протекает очень медленно), для определения восстановителей используют метод обратного титрования. Для этого нужны два рабочих раствора — йода и тиосульфата натрия. К определяемому восстановителю добавляют точно отмеренный объем раствора йода, взятого в избытке по отношению к восстановителю. Между йодом и восстановителем происходит реакция, затем остаток непрореагировавшего йода оттитровывается раствором тиосульфата. Например, при определении H2S: H2S + I2 = 2I- + S + 2H+; Избыток I2 + 2 S2O32- = 2I- + S4O2-6 Остаток Этим методом определяют большие концентрации H2S, сульфидов, сульфитов, ряда металлов в порошках (например, цинка), некоторых органических соединений. Для определения окислителей методом заместительного титрования поступают следующим образом. К подкисленному серной кислотой раствору KI, взятому в избытке, прибавляют точно отмеренный пипеткой Мора объем раствора определяемого окислителя (например, КСlО3) и выделившийся йод оттитровывают тиосульфатом натрия: СlO-3 +6I- + 6Н+ = Сl- + 3I2 + 3Н2О; I2 + 2S4O2-6 Число молей эквивалентов Na2S2O3 равно числу молей эквивалентов йода, а последнее равно числу молей эквивалентов определяемого окислителя (КСlO3). Таким образом, хотя определяемый окислитель и Na2S2O3 непосредственно друг с другом не реагируют, тем не менее их количества эквивалентны. Поэтому для вычисления можно пользоваться обычной формулой, применяемой при прямом титровании: С(1/6КСlO3) • V(КСlO3) = C(Na2S2O3) • V(Na2S2O3). Этим методом определяют многие окислители, например Сl2, Вr2, КМnО4, СаОСl2, нитриты, Н2О2, соли Fe(III), Cu(II), соединения мышьяка (V). Кристаллический йод обычно загрязнен соединениями хлора и брома, содержит влагу, поэтому его очищают сублимацией из смеси с KI и СаО. Из-за своей летучести кристаллический йод не является исходным веществом, и его редко используют в качестве первичного стандарта. Чаще всего сначала готовят раствор йода с концентрацией, приблизительно равной требуемой, а затем точную концентрацию раствора устанавливают с помощью стандартизированного раствора тиосульфата натрия. Молекулярный йод плохо растворяется в воде. Для приготовления раствора пользуются хорошей растворимостью йода в растворе KI вследствие образования комплексного соединения: I2 + KI ↔ К[I3]. Трийодид-ион J-3 и молекулярный йод в ОВ-реакциях ведут себя одинаково, поэтому при йодометрических определениях соответствующие уравнения реакций записывают с участием I2, а не иона J-3 . Раствор тиосульфата натрия готовят из кристаллогидрата Na2S2O3 • 5Н2О, состав которого не бывает строго постоянным. Кроме того, при растворении тиосульфата натрия происходит частичное разложение его растворенным в воде СО2: Na2S2O3 + СО2 + Н2O = NaHCO3 + NaHSO3 + S↓. С другой стороны, тиосульфат-ионы медленно реагируют с молекулярным кислородом воздуха: 2 S2O2-3 + О2 = 2 SO2-4 + 2S↓. Из-за выделения серы раствор сначала мутнеет, а затем на дне сосуда собирается белый осадок. Чтобы уменьшить разложение тиосульфат-иона, для приготовления раствора используют свежепрокипяченную охлажденную воду. Приготовленный раствор оставляют на 8—10 дней, после чего приступают к определению его концентрации по первичному стандарту. В качестве первичных стандартов для определения концентрации раствора Na2S2O3 служат дихромат калия К2Сr2O7 или йодат калия КIО3, из которых по навеске легко получить раствор с известной концентрацией. Следует отметить, что тиосульфат натрия является сильным восстановителем (E0(S4O2-6 /2 S2O2-3) = + 0,08 В), реагирует быстро и стехиометрично с образованием тетратионат-ионов только с трийодид-ионами. Со многими другими окислителями, например с МnO-4 , Сr2О2-7 , ВrО-3, IО-3 и т.д., тиосульфат-ионы реагируют нестехиометрично. Это исключает возможность использования прямого титрования окислителей раствором Na2S2O3. Установление точной концентрации раствора Na2S2O3 с использованием в качестве первичного стандарта дихромата или йодата калия основано на методе косвенного (заместительного) титрования. 3. Рефрактометрия қандай қосылыстарды талдау үшін қолданылады? Интенсивное развитие рефрактометрии как инструментального метода химических исследований началось со второй половины Х1Х века, когда одной из центральных проблем химии стало выяснение зависимости свойств веществ от их состава и строения, а быстро растущая химическая промышленность потребовала разработки удобных и простых методов технического анализа. Особая роль рефрактометрии в производстве оптического стекла предопределила то внимание, которое уделялось ей при создании оптической промышленности России. Одной из первых проблем Государственного оптического института было систематическое исследование свойств оптических материалов. В связи с этим тщательно изучались и усовершенствовались различные способы точных измерений показателей преломления и их термических коэффициентов. Для производственного контроля оптических стекол, измерения их дисперсии в крайних областях спектра и в пределах полос поглощения были созданы уникальные установки и разработаны оригинальные конструкции специальных приборов. Широкому распространению рефрактометрии способствовало исключительно ценное совмещение высокой точности, технической простоты и доступности. Показатель преломления принадлежит к числу немногих физических констант, которые можно измерить с очень высокой точностью и небольшой затратой времени, располагая лишь малым количеством вещества. Измерение показателя преломления дает возможность непосредственно установить концентрацию двухкомпонентных растворов. Для этого используются эмпирические расчетные формулы и графики. Сочетание рефрактометрических измерений с определением других физических свойств или с химической обработкой исследуемого вещества позволяет анализировать тройные и более сложные смеси и определять, таким образом, состав многих важных промышленных продуктов и биологических объектов. Кроме самого показателя преломления, в химии используется ряд более сложных функций, к которым относят различные виды формул удельной и молекулярной рефракции. Каждая из этих величин имеет свои особенности, которые должны учитываться при ее практическом использовании. Так, рефрактометрический анализ двойных систем основывается на употреблении показателя преломления, а применение для этой цели рефракционной дисперсии или удельной рефракции практически бесполезно. В тоже время дисперсия и удельная рефракция с успехом используются в анализе сложных углеводородных смесей, где измерение одного только показателя преломления недостаточно. Показатель преломления служит важным критерием чистоты вещества, но молекулярная рефракция и дисперсия для этой цели мало пригодны. Однако для рефрактометрического определения строения органических соединений именно эти последние костанты очень удобны. Молекулярная рефракция непосредственно связана с поляризуемостью ионов и молекул, то есть со способностью их электронных оболочек деформироваться во внешнем электрическом поле или электрическом поле других ионов и молекул. Деформируемость электронных оболочек, а, следовательно, и молекулярная рефракция являются важным критерием, характеризующим многие физические и химические свойства вещества. У органических соединений обнаруживается закономерное изменение молекулярной рефракции и дисперсии в зависимости от состава и строения в пределах гомологических рядов. Эти закономерности позволяют использовать рефрактометрию для классификации и определения строения органических соединений. Благодаря простоте и доступности рефрактометрические методы сохранили свое значение в химии до настоящего времени, несмотря на бурное развитие молекулярной спектроскопии, и других физических методов определения строения органических соединений. Но следует учитывать, что применение рефрактометрических методов оправдывается лишь в тех случаях, когда они дают не только наиболее простое, но и достаточно обоснованное однозначное решение поставленной задачи. 4. Рефрактометриядағы концентрацияны есептеу қалай жүргізіледі? В рефрактометрии используют два способа расчета концентрации вещества в растворе по измеренному показателю преломления. 1. Расчет концентрации по формуле. Зависимость показателя преломления от концентрации вещества в процентах выражается: n=n0+С·F, отсюда 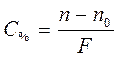 (1), (1), где n и n0 – показатели преломления раствора и растворителя; F – фактор показателя преломления. 2. Расчет концентрации по рефрактометрическим таблицам. Измерив показатель преломления, в таблице находят соответствующее ему значение концентрации. Если измеренный показатель преломления в таблице не приведен, проводится интерполирование. Рефрактометрия – метод анализа, основанный на измерении показателя (коэффициента) преломления света (n) при прохождении его из одной среды в другую. Преломление света, то есть изменение его первоначального направления, обусловлено различной скоростью распределения света в различных средах. При этом отношение синуса угла падения луча (α) к синусу угла преломления (β) для двух соприкасающихся сред есть величина постоянная, называемая показателем преломления (n). Показатель преломления также равен отношению скоростей распространения света в этих средах:  В лабораторных условиях обычно определяют так называемый относительный показатель преломления вещества по отношению к воздуху помещения, где ведется измерение. Показатель преломления измеряют на приборах рефрактометрах различных систем. Величина показателя преломления зависит от факторов, влияющих на скорость распространения света в испытуемом веществе (природы вещества, длины волны света, температуры, при которой проводится измерение, концентрации вещества в растворе) и среды, относительно которой он измеряется. Измерение показателя преломления проводят при длине волны света 589,3 нм (линия D спектра натрия). Обязательным условием определения показателя преломления является соблюдение температурного режима. Обычно определение выполняется при 20±0,30С. При повышении температуры величина показателя преломления уменьшается, при понижении – увеличивается. Поправку рассчитывают по следующей формуле:n1=n20+ (20-t)·0,0002 Диапазон значений измеряемого показателя преломления 1,3 - 1,7. Точность измерения +2·10-4. Величина прироста показателя преломления при увеличении концентрации на каждый процент называется фактором показателя преломления (F). Значение F устанавливают экспериментально для каждого вещества и каждого процента концентрации. При концентрации вещества менее 3 – 4% не рекомендуется использовать метод рефрактометрии для количественного определения веществ. В лабораторных условиях обычно определяют так называемый относительный показатель преломления вещества по отношению к воздуху помещения, где ведется измерение. Показатель преломления измеряют на приборах рефрактометрах различных систем. Величина показателя преломления зависит от факторов, влияющих на скорость распространения света в испытуемом веществе (природы вещества, длины волны света, температуры, при которой проводится измерение, концентрации вещества в растворе) и среды, относительно которой он измеряется. Измерение показателя преломления проводят при длине волны света 589,3 нм (линия D спектра натрия). Обязательным условием определения показателя преломления является соблюдение температурного режима. Обычно определение выполняется при 20±0,30С. При повышении температуры величина показателя преломления уменьшается, при понижении – увеличивается. Поправку рассчитывают по следующей формуле:n1=n20+ (20-t)·0,0002 Диапазон значений измеряемого показателя преломления 1,3 - 1,7. Точность измерения +2·10-4. Величина прироста показателя преломления при увеличении концентрации на каждый процент называется фактором показателя преломления (F). Значение F устанавливают экспериментально для каждого вещества и каждого процента концентрации. При концентрации вещества менее 3 – 4% не рекомендуется использовать метод рефрактометрии для количественного определения веществ. 5. Фармацевтикалық талдауда жоғары өнімді сұйықтық хроматографиясы не үшін қолданылады? Высокоэффективная жидкостная хроматография (жидкостная хроматография высокого давления) – это метод колоночной хроматографии, в котором подвижной фазой служит жидкость, движущаяся через хроматографическую колонку, заполненную неподвижной фазой (сорбентом). Колонки для высокоэффективной жидкостной хроматографии характеризуются высоким гидравлическим сопротивлением на входе. В зависимости от механизма разделения веществ различают следующие варианты высокоэффективной жидкостной хроматографии: адсорбционную, распределительную, ионообменную, эксклюзионную, хиральную и др. в соответствии с характером основных проявляющихся межмолекулярных взаимодействий. В адсорбционной хроматографии разделение веществ происходит за счет их различной способности адсорбироваться и десорбироваться с поверхности сорбента с развитой поверхностью, например, силикагеля. В распределительной высокоэффективной жидкостной хроматографии разделение происходит за счет различия коэффициентов распределения разделяемых веществ между неподвижной (как правило, химически привитой к поверхности неподвижного носителя) и подвижной фазами. В зависимости от типа подвижной и неподвижной фазы различают нормально-фазовую и обращенно-фазовую хроматографию. В нормально-фазовой высокоэффективной жидкостной хроматографии неподвижная фаза – полярная (чаще всего силикагель или силикагель с привитыми NH2— или CN-группами и др.), а подвижная фаза – неполярная (гексан, либо смеси гексана с более полярными органическими растворителями – хлороформом, спиртами и т.д.). Удерживание веществ растет с увеличением их полярности. В нормально-фазовой хроматографии элюирующая способность подвижной фазы увеличивается с ростом ее полярности. В обращенно-фазовой хроматографии неподвижная фаза – неполярная (гидрофобные силикагели с привитыми группами С4, С8, С18 и др.); подвижная фаза – полярная (смеси воды и полярных растворителей: ацетонитрила, метанола, тетрагидрофурана и др.). Удерживание веществ растет с увеличением их гидрофобности (неполярности). Чем больше содержание органического растворителя, тем выше элюирующая способность подвижной фазы. В ионообменной хроматографии молекулы веществ смеси, диссоциированные в растворе на катионы и анионы, разделяются при движении через сорбент (катионит или анионит) за счет различной силы взаимодействия определяемых ионов с ионными группами сорбента. В эксклюзионной (ситовой, гель-проникающей, гель-фильтрационной) хроматографии молекулы веществ разделяются по размеру за счет их разной способности проникать в поры неподвижной фазы. При этом первыми из колонки выходят наиболее крупные молекулы, способные проникать в минимальное число пор неподвижной фазы, а последними выходят вещества с малыми размерами молекул. В хиральной хроматографии происходит разделение оптически активных соединений на отдельные энантиомеры. Разделение может осуществляется на хиральных неподвижных фазах или на ахиральных неподвижных фазах с использованием хиральных подвижных фаз. Существуют и другие варианты высокоэффективной жидкостной хроматографии. Часто разделение протекает не по одному, а по нескольким механизмам одновременно, в зависимости от типа подвижной и неподвижной фаз, а также природы определяемого соединения. Область применения. Высокоэффективная жидкостная хроматография успешно применяется как для качественного, так и для количественного анализа лекарственных средств в испытаниях «Подлинность», «Посторонние примеси», «Растворение», «Однородность дозирования», «Количественное определение». Следует отметить, что хроматография позволяет совмещать в одной пробе несколько испытаний, в том числе «Подлинность» и «Количественное определение». Қолданылған әдебиеттер тізімі Фармацевтическая химия: учебное пособие / под ред. А.П. Арзамасцева. - 3 -е изд., испр. - М.: ГЭОТАР - Медиа, 2006. 17. Фармацевтическая химия / Глущенко Н.Н., Плетнева Т.В., Попков В.А. a. - М., 2004.- 382с. . Адсорбционная хроматография Лабораторное пособие под ред. Пол С. Садек. - "Как избежать ошибок в Высокоэффективной Жидкостной Хроматографии." - М.,2000. - 447с. . Высокоэффективная жидкостная хроматография: Основы теории. Методология. Применение в лекарственной химии / под ред. В.Д. Шатц - М., 2006. - 267с. . Высокоэффективная жидкостная хроматография на микроколоночных хроматографах серии «Милихром» / под ред. С.Н. Сычев - М.,2000. - 345с. . Сычев С.Н., Сычев К.С., Гаврилина В.А. "Высокоэффективная жидкостная хроматография на микроколоночных хроматографах серии "Миллихром" Орел 2002 / С.Н. Сычев - 2002. - 656с. . Рудаков О.Б. Методы жидкостной хроматографии / О.Б. Рудаков. - 2007. - С. 234. . Бражников В.В. Детекторы для хроматографии / В.В. Бражников. 2004. - С. 241. . Васильев В.П. Аналитическая химия. Кн. 1: Титриметрические и гравиметрический методы анализа. - М.: Дрофа, 2005. - 366 с. Васильев В.П. Аналитическая химия. Лабораторный практикум.- М.: Дрофа, 2004.- 416 с. Васильев В.П. Аналитическая химия. Сборник вопросов, упражнений и задач.- М.: Дрофа, 2004. - 318 с. Харитонов Ю.Я. Аналитическая химия (аналитика): учебник для вузов. В 2 кн. Кн. 2. Количественный анализ. Физико-химические (инструментальные) методы анализа. – М.: Высшая школа, 2001. - 559 с. |