коллок по био. # 3коллоквиум по био. 3. Роль ядра в передаче наследственных признаков. Опыты Астаурова по андрогенезу
 Скачать 105.17 Kb. Скачать 105.17 Kb.
|

|
3 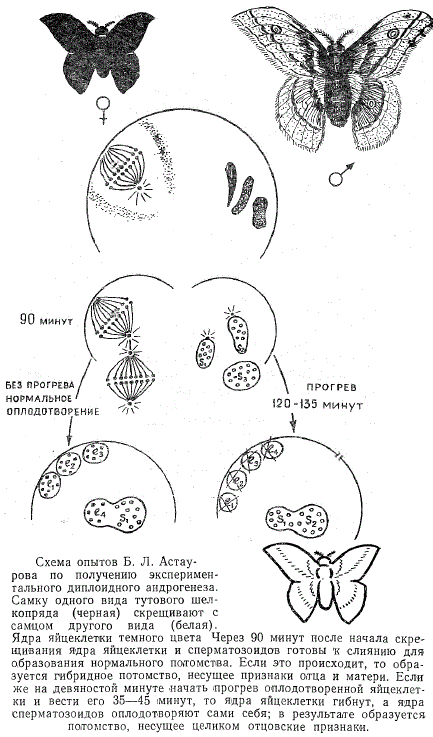 . Роль ядра в передаче наследственных признаков. Опыты Астаурова по андрогенезу. . Роль ядра в передаче наследственных признаков. Опыты Астаурова по андрогенезу.Роль ядра: участие в делении клетки, хранение и передача наследственных признаков организма, регуляция процессов жизнедеятельности в клетке, благодаря генетической информации, записанной в молекуле ДНК. В ядре каждой клетки содержится основная наследственная информация, необходимая для развития целого организма со всем разнообразием его свойств и признаков. Именно ядро играет центральную роль в явлениях наследственности. Андрогенез-развитие яйцеклетки с мужским ядром, привнесённым в неё спермием в процессе оплодотворения. Опыт проводился на тутовом шелкопряде. Он хотел получить андрогенное потомство. Межвидовое скрещивание двух шелкопрядов. Через некоторое время происходит воздействие температурой на яйцо оплодотворённой самки. Ядра сперматозоидов проходят в середину яйцеклетки и ждут слияния Но как результат нагревания, ядро яйцеклетки погибает как пишет Астауров, «мужским гаплоидным ядрам не остается ничего другого, как копулировать между собой», то есть совершать «своеобразное мужское самооплодотворение». Образуется потомство несущее целиком отцовские признаки. 4. Особенности молекулярного строения ДНК И РНК. Модель структуры ДНК Уотсона крика. Правило чаргаффа. Комплиментарность структуры ДНК. Проблема избыточности ДНК. Среди нуклеиновых кислот различают два вида соединений: дезоксирибонуклеиновую (ДНК) и рибонуклеиновую (РНК) кислоты. Наиболее химически устойчивым компонентом является ДНК, которая представляет собой субстрат наследственности и изменчивости. ДНК состоит из нуклеотидов, в состав которых входят сахар — дезоксирибоза, фосфат и одно из азотистых оснований — пурин (аденин или гуанин) либо пиримидин (тимин или цитозин). Особенностью структурной организации ДНК является то, что ее молекулы включают две полинуклеотидные цепи, связанные между собой определенным образом. Цепи соединяются друг с другом водородными связями между их азотистыми основаниями по принципу комплементарности. Аденин одной цепи соединяется двумя водородными связями с тимином другой цепи, а между гуанином и цитозином разных цепей образуются три водородные связи. Такое соединение азотистых оснований обеспечивает прочную связь двух цепей и сохранение равного расстояния между ними на всем протяжении. Другой важной особенностью объединения двух полинуклеотидных цепей в молекуле ДНК является их антипараллельность: 5'-конец одной цепи соединяется с 3'-концом другой, и наоборот. Данные рентгеноструктурного анализа показали, что молекула ДНК, состоящая из двух цепей, образует спираль, закрученную вокруг собственной оси. Диаметр спирали составляет 2 нм, длина шага — 3, 4 нм. В каждый виток входит 10 пар нуклеотидов. Чаще всего двойные спирали являются правозакрученными — при движении вверх вдоль оси спирали цепи поворачиваются вправо. Большинство молекул ДНК в растворе находится в правозакрученной Таким образом, в структурной организации молекулы ДНК можно выделить первичную структуру —полинуклеотидную цепь, вторичную структуру—две комплементарные друг другу и антипараллельные полинуклеотидные цепи, соединенные водородными связями, и третичную структуру — трехмерную спираль. Трехмерная модель пространственного строения двухцепочечной ДНК была описана в 1953 г. Дж. Уотсоном и Френсисом Криком. Согласно этой модели молекула ДНК состоит из двух полинуклеотидных цепей, которые образуют правую спираль (винтовую линию) относительно одной и той же оси. Направление цепей взаимно противоположное. Еще до открытия Уотсона и Крика, в 1950 г. австралийский биохимик Эдвин Чаргафф установил, что в ДНК любого организма количество адениловых нуклеотидов равно количеству тимидиловых, а количество гуаниловых нуклеотидов равно количеству цитозиловых нуклеотидов (А=Т, Г=Ц), илисуммарное количество пуриновых азотистых оснований равно суммарному количеству пиримидиновых азотистых оснований (А+Г=Ц+Т). Эти закономерности получили название «правила Чаргаффа».Обращает на себя внимание явная избыточность кода, проявляющаяся в том, что многие аминокислоты шифруются несколькими триплетами. Это свойство триплетного кода, названное вырожденностью, имеет очень важное значение, так как возникновение в структуре молекулы ДНК изменений по типу замены одного нуклеотида в полинуклеотидной цепи может не изменить смысла триплета. Возникшее таким образом новое сочетание из трех нуклеотидов кодирует ту же самую аминокислоту. 5. Современные представления о генетическом коде, опыты ниринберга, репликация, транскрипция, трансляция. Транскрипция 4 значного кода первичной генетической информации в 20 значный аминокислотный код белков. Мультимерная организация белков ( гемоглобин человека) Последовательность аминокислот в пептидах зашифрована в молекулах ДНК с помощью биологического (генетического) кода. 1. ТРИПЛЕТНОСТЬ. Кодирование информации в молекулах ДНК должно осуществляться сочетаниями нескольких нуклеотидов. В многообразии белков, существующих в природе, было обнаружено около 20 различных аминокислот. Для шифровки такого их числа достаточное количество сочетаний нуклеотидов может обеспечить лишь триплетный код, в котором каждая аминокислота шифруется тремя стоящими рядом нуклеотидами. В этом случае из четырех нуклеотидов образуется 4(в 3 степени) = 64 триплета. Из 64 возможных триплетов ДНК 61 кодирует различные аминокислоты; оставшиеся 3 получили название бессмысленных, или «нонсенстриплетов». Они не шифруют аминокислот и выполняют функцию знаков препинания при считывании наследственной информации. К ним относятся АТТ, АЦТ, АТЦ. 2. ВЫРОЖДЕННОСТЬ И ИЗБЫТОЧНОСТЬ. Обращает на себя внимание явная избыточность кода, проявляющаяся в том, что многие аминокислоты шифруются несколькими триплетами . Это свойство триплетного кода, названное вырожденностью, имеет очень важное значение, так как возникновение в структуре молекулы ДНК изменений по типу замены одного нукле-отида в полинуклеотидной цепи может не изменить смысла триплета. Возникшее таким образом новое сочетание из трех нуклеотидов кодирует ту же самую аминокислоту. 3. СПЕЦИФИЧНОСТЬ. В процессе изучения свойств генетического кода была обнаружена его специфичность. Каждый триплет способен кодировать только одну определенную аминокислоту. 4. УНИВЕРСАЛЬНОСТЬ. Интересным фактом является полное соответствие кода у различных видов живых организмов. Такая универсальность генетического кода свидетельствует о единстве происхождения всего многообразия живых форм на Земле в процессе биологической эволюции. Про отличия у митохондрий. Незначительные отличия генетического кода обнаружены в ДНК митохондрий некоторых видов. Это не противоречит в целом положению об универсальности кода, но свидетельствует в пользу определенной дивергентности в его эволюции на ранних этапах существования жизни. Расшифровка кода в ДНК митохондрий различных видов показала, что во всех случаях в митохондриальных ДНК отмечается общая особенность: триплет АЦТ читается как АЦЦ, и поэтому из нонсенс-триплета превращается в шифр аминокислоты триптофана. К 1961 году стало ясно, что код триплетный, вырожденный и неперекрывающийся (то есть считывание происходит кодон за кодоном) и что он содержит инициирующие и терминирующие кодоны. Расшифровка генетического кода усложнялась тем, что в составе ДНК, как было известно, имеется только 4 типа нуклеотидов: адениловые (А), гуаниловые (Г), тимидиловые (Т) и цитидиловые (Ц), а в составе белков — 20 основных аминокислот. Теоретич. анализ решения этой задачи был предпринят в 1954 г. амер. физиком Г. Гамовым, к-рый предположил, что каждую аминокислоту кодирует тройка нуклеотидов — так наз. триплет, или кодон. Таких кодонов должно было быть 64 (число сочетаний четырех элементов в группах по три равно 43, или 64), а это более чем в три раза превышает число основных аминокислот в белке. В связи с этим было высказано предположение, что одной аминокислоте может соответствовать не один, а несколько кодонов. Дело было лишь за тем, чтобы установить соответствие каждого аминокислотного остатка конкретным кодонам и узнать, какие кодоны обозначают начало и конец синтеза белковой цепи. Именно этим принципом дешифровки кода и воспользовались М. Ниренберг и Дж. Маттеи в 1961 году. К этому времени как раз научились синтезировать искусственные РНК. Живая клетка такую РНК расщепит до отдельных нуклеотидов, а их использует для строительства собственных РНК. Поэтому Ниренберг и Маттеи использовали не живые клетки, а клеточные экстракты, которые сохраняли способность синтезировать белок на РНК, но не содержали ферментов, расщепляющих РНК. Такие экстракты назвали бесклеточной системой. Ниренберг и Маттеи получили экстракт из кишечной палочки и добавили к нему гомополимер, состоящий только из урацилов. Так бесклеточной системе был задан первый вопрос: какой аминокислоте соответствует кодон УУУ? Ответ был однозначен: кодону УУУ отвечает фенилаланин. Этот ответ произвел настоящую сенсацию. Путь к расшифровке кода был открыт. (КРАТКО: Опыт Ниренберга: он создал искусственную иРНК и поместил ее в бесклеточную среду, содержащую аминокислоты, РНК, все необходимое для синтеза белка. В результате многочисленых опытов, он заметил ычто происходил синтез только фенилаланина = триплет УУУ, так открыт был 1-ый триплет. Одним из центральных процессов метаболизма клетки, свящанных с потоком в-ва является синтез белка – формирование сложной молекулы белка-полимера из аминокислот-мономеров. Процесс протекает так: ДНК, затем РНК, затем БЕЛОК) Репликация ДНК. Одним из основных свойств материала наследственности является его способность к самокопированию — репликация. Это свойство обеспечивается особенностями химической организации молекулы ДНК, состоящей из двух комплементарных цепей. В процессе репликации на каждой полинуклеотидной цепи материнской молекулы ДНК синтезируется комплементарная ей цепь. В итоге из одной двойной спирали ДНК образуются две идентичные двойные спирали. Такой способ удвоения молекул, при котором каждая дочерняя молекула содержит одну материнскую и одну вновь синтезированную цепь, называют полуконсервативным (Более подробно можно почитать на стр 72, 2003 ТРАНСКРИПЦИЯ - процесс синтеза м РНК. Синтез мРНК начинается с обнаружения РНК-полимеразой особого участка в молекуле ДНК, который указывает место начала транскрипции — промотора. После присоединения к промотору РНК-полимераза раскручивает прилежащий виток спирали ДНК. Две цепи ДНК в этом месте расходятся, и на одной из них фермент осуществляет синтез мРНК. Сборка рибонуклеотидов в цепь происходит с соблюдением их комплементарности нуклеотидам ДНК, а также антипараллельно по отношению к матричной цепи ДНК. В связи с тем, что РНКполимераза способна собирать полинуклеотид лишь от 5'-конца к 3'-концу, матрицей для транскрипции может служить только одна из двух цепей ДНК, а именно та, которая обращена к ферменту своим 3'-концом (3' → 5'). Такую цепь называют кодогенной. Антипараллельность соединения двух полинуклеотидных цепей в молекуле ДНК позволяет РНК-полимеразе правильно выбрать матрицу для синтеза мРНК. Продвигаясь вдоль кодогенной цепи ДНК, РНК-полимераза осуществляет постепенное точное переписывание информации до тех пор, пока она не встречает специфическую нуклеотидную последовательность — терминатор транскрипции. В этом участке РНК-полимераза отделяется как от матрицы ДНК, так и от вновь синтезированной мРНК. Фрагмент молекулы ДНК, включающий промотор, транскрибируемую последовательность и терминатор, образует единицу транскрипции — транскриптон. В процессе синтеза, по мере продвижения РНК-полимеразы вдоль молекулы ДНК, пройденные ею одноцепочечные участки ДНК вновь объединяются в двойную спираль. Образуемая в ходе транскрипции мРНК содержит точную копию информации, записанной в соответствующем участке ДНК. Тройки рядом стоящих нуклеотидов мРНК, шифрующие аминокислоты, называют кодонами. Последовательность кодонов мРНК шифрует последовательность аминокислот в пептидной цепи. Кодонам мРНК соответствуют определенные аминокислоты. ТРАНСЛЯЦИЯ. Наследственная информация, «записанная» в молекулах ДНК и «переписанная» на мРНК, расшифровывается в ходе трансляции благодаря двум процессам специфического узнавания молекулярных поверхностей. Сначала фермент аминоацил-тРНК-синтетаза обеспечивает соединение тРНК с транспортируемой ею аминокислотой. Затем аминоацил-тРНК комплементарно спаривается с мРНК благодаря взаимодействию антикодона с кодоном. С помощью системы тРНК язык нуклеотидной цепи мРНК. транслируется в язык аминокислотной последовательности пептида. (На странице 99 2003 г можно про молекулу тРНК почитать) Мультимерные белки - белки содержащие более чем одну субъединицу. Если субъединицы белка одинаковы, то белок-гомомультимер, детерминируемый одним геном. Если же субъединицы белка различны, то белок называют гетеромультимером. Гемоглобин служит примером белка , состоящего более чем из одного типа субъединиц. Группа гема связана с двумя а-субъединицами двумя р-субъединица-ми. Каждый тип субъединиц иная полипептидная цепь и продукт другого гена. Таким образом , функция гемоглобина может быть подавлена мутацией в любом из генов, кодирующих либо а-, либо Р-субъединиц. 9. Хромосомная теория наследственности Т. Моргана, ее основные положения. Группы сцепленных генов. Термин хромосома был предложен в 1888 г. немецким морфологом В. Вальдейером, который применил его для обозначения внутриядерных структур эукариотической клетки, хорошо окрашивающихся основными красителями (от греч. хрома — цвет, краска, и сома — тело). К началу XX в. углубленное изучение поведения этих структур в ходе самовоспроизведения клеток, при созревании половых клеток, при оплодотворении и раннем развитии зародыша обнаружило строго закономерные динамические изменения их организации. Это привело немецкого цитолога и эмбриолога Т. Бовери (1902—1907) и американского цитолога У. Сеттона (1902—1903) к утверждению тесной связи наследственного материала с хромосомами, что легло в основу хромосомной теории наследственности. Детальная разработка этой теории была осуществлена в начале XX в. школой американских генетиков, возглавляемой Т. Морганом. Работы Т. Моргана и его сотрудников не только подтвердили значение хромосом как основных носителей наследственного материала, представленного отдельными генами, но и установили линейность расположения их по длине хромосомы. Доказательством связи материального субстрата наследственности и изменчивости с хромосомами было, с одной стороны, строгое соответствие открытых Г. Менделем закономерностей наследования признаков поведению хромосом в ходе митоза, при мейозе и оплодотворении. С другой стороны, в лаборатории Т. Моргана был обнаружен особый тип наследования признаков, который хорошо объяснялся связью соответствующих генов с Х-хромосомой. Речь идет о сцепленном с полом наследовании окраски глаз у дрозофилы. Представление о хромосомах как носителях комплексов генов было высказано на основе наблюдения сцепленного наследования ряда родительских признаков друг с другом при передаче их в ряду поколений. Такое сцепление неальтернативных признаков было объяснено размещением соответствующих генов в одной хромосоме, которая представляет собой достаточно устойчивую структуру, сохраняющую состав генов в ряду поколений клеток и организмов. Согласно хромосомной теории наследственности, совокупность генов, входящих в состав одной хромосомы, образует группу сцепления. Каждая хромосома уникальна по набору заключенных в ней генов. Число групп сцепления в наследственном материале организмов данного вида определяется, таким образом, количеством хромосом в гаплоидном наборе их половых клеток. При оплодотворении образуется диплоидный набор, в котором каждая группа сцепления представлена двумя вариантами — отцовской и материнской хромосомами, несущими оригинальные наборы аллелей соответствующего комплекса генов. Представление о линейности расположения генов в каждой хромосоме возникло на основе наблюдения нередко возникающей рекомбинации (взаимообмена) между материнским и отцовским комплексами генов, заключенными в гомологичных хромосомах. Было установлено, что частота рекомбинации характеризуется определенным постоянством для каждой пары генов в данной группе сцепления и различна для разных пар. Это наблюдение дало возможность высказать предположение о связи частоты рекомбинации с последовательностью расположения генов в хромосоме и процессом кроссинговера, происходящим между гомологами в профазе I мейоза. Представление о линейном распределении генов хорошо объясняло зависимость частоты рекомбинации от расстояния между ними в хромосоме. Открытие сцепленного наследования неальтернативных признаков легло в основу разработки методики построения генетических карт хромосом с использованием гибридологического метода генетического анализа. Таким образом, в начале XX в. была неопровержимо доказана роль хромосом как основных носителей наследственного материала в эука-риотической клетке. Подтверждение этому было получено при изучении химического состава хромосом. Основные положения хромосомной теории наследственности: 1.Гены расположены в хромосомах линейно в определенных участках – локусах. Аллельные гены занимают одинаковые локусы гомологичных хромосом. 2.Гены, расположенные в одной хромосоме, образуют группу сцепления и наследуются вместе или сцеплено. Число групп сцепления = числу хромосом в гаплоидном наборе. 3.Между гомологичными хромосомами возможен кроссинговер, нарушающий сцепление. 4.процесс кроссинговера прямо пропорционален расстоянию между генами. 10. Хромосомное определение пола. Сцепленное с полом наследование (X-сцепленное и голандрическое-Y наследование). Сцепленное с полом наследование. Анализ наследования признака окраски глаз у дрозофилы в лаборатории Моргана выявил некоторые особенности, заставившие выделить в качестве отдельного типа наследования признако сцепленное с полом наследовании. Ген, определяющий окраску глаз у дрозофилы, расположен в Х-хромосоме и не имеет гомолога в Y-хромосоме. Все особенности сцепленного с полом наследования объясняются неодинаковой дозой соответствующих генов у представителей разного — гомо- и гетерогаметного пола. Гомогаметный пол несет двойную дозу генов, расположенных в Х-хромосоме. Гетерогаметный пол имеет одну Х- хромосому (ХО или XY). У некоторых видов Y-хромосома генетически инертна, у других она содержит некоторое количество структурных генов, часть из которых гомологична генам Х-хромосомы. Гены негомологичных участков у гетерогаметного пола находятся в гемизиготном состоянии: ХAY, ХaХ, XYB. Формирование таких признаков у гетерогаметного пола определяется тем, какой аллель данного гена присутствует в генотипе организма. Характер наследования сцепленных с полом признаков в ряду поколений зависит от того, в какой хромосоме находится соответствующий ген. Х-сцепленное наследование. Х-хромосома присутствует в кариотипе каждой особи, поэтому признаки, определяемые генами этой хромосомы, формируются у представителей как женского, так и мужского пола. Особи гомогаметного пола получают эти гены от обоих родителей и через свои гаметы передают их всем потомкам. Мужской пол получает Х- сцепленные гены от матери и передает их дочерям (при этом муж.пол никогда не наследует отцовского Х-хр-му и не передает его своим сыновьям). Так как у гомогаметного пола признак развивается в результате взаимодействия аллельных генов, различают Х-сцепленное доминантное и Х-сцепленное рецессивное наследование. Х-сцепленный доминантный признак (красный цвет глаз у дрозофилы) передается самкой всему потомству. Самец передает свой Х-сцепленный доминантный признак лишь самкам следующего поколения. Самки могут наследовать такой признак от обоих родителей, а самцы — только от матери. Х-сцепленный рецессивный признак, (белый цвет глаз у дрозофилы) у самок проявляется только при получении ими соответствующего аллеля от обоих родителей (XaXa). У самцов XaY он развивается при получении рецессивного аллеля от матери. Рецессивные самки передают рецессивный аллель потомкам любого пола, а рецессивные самцы —только «дочерям». При Х-сцепленном наследовании, так же как и при аутосомном, возможен промежуточный характер проявления признака у гетерозигот. Например, у кошек пигментация шерсти контролируется Х-сцепленным геном, разные аллели которого определяют черную XA и рыжую XA’ пигментацию. Гетерозиготные самки XAXA имеют пеструю окраску шерсти. Самцы же могут быть либо черными (XAY, либо рыжими (XA’Y). Голандрическое наследование. Активно функционирующие гены Y-хромосомы, присутствуют в генотипе только гетерогаметного пола, причем в гемизиготном состоянии. Поэтому они проявляются фенотипически и передаются из поколения в поколение лишь у представителей гетерогаметного пола. Так, у человека признак гипертрихоза ушной раковины («волосатые уши») наблюдается исключительно у мужчин и наследуется от отца к сыну. 11. Половой хроматин и его значение в выявлении хромосомных болезней: Половой хроматин — особые хроматиновые тельца клеточных ядер особей женского пола у человека и других млекопитающих. Располагаются у ядерной оболочки, на препаратах имеют обычно треугольную или овальную форму. Половой хроматин изучают при цитологическом определении пола, для выявления хромосомных болезней. Половой хроматин изучают при цитологическом определении пола; для выявления хромосомных болезней (синдромом Шершевского - Тёрнера, для которого характерно отсутствие полового хроматина у женщин; синдром Клайнфелтера, при котором у мужчин выявляют половой хроматин; синдром трисомии X, при котором в ядре вместо одного тельца полового хроматина выявляют два), в судебной медицине для установления принадлежности пола. В качестве экспресс-метода, выявляющего изменение числа половых хромосом, используют метод определения полового хроматина в неделящихся клетках слизистой оболочки щеки. Половой хроматин, или тельце Барра, образуется в клетках женского организма одной из двух Х-хромосом. Оно выглядит как интенсивно окрашенная глыбка, расположенная у ядерной оболочки. При увеличении количества Х-хромосом в кариотипе организма в его клетках образуются тельца Барра в количестве на единицу меньше числа Х-хромосом. При уменьшении числа Х-хромосом (моносомия X) тельце Барра отсутствует. В мужском кариотипе Y-хромосома может быть обнаружена по более интенсивной по сравнению с другими хромосомами люминесценции при обработке их акрихинипритом и изучении в ультрафиолетовом свете. Синдром Клайнфельтера — наследственное заболевание. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола. Наиболее распространён синдром Клайнфельтера (47, XXY). Клиника: · Неразвитые половые железы · Часто умственная отсталость · Высокий рост · Можно увидеть тельца Барра Тельца Барра - это Х-половой хроматин, свернутый в плотную гетерохроматидную структуру, неактивный. Синдром Шерешевского — Тёрнера — хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом. Моносомия по X-хромосоме (XО)(45 хромосом). Причина: нерасхождение половых хромосом. Клиника: · Всегда страдают девочки · Низкий рост (карликовость) · Умственная отсталость · Монголоидный разрез глаз · «шея сфинкса» XYY-синдром, также известен как YY-синдром или Синдром Джейкобса — хромосомное заболевание, характерное только для мужчин. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Внешне мужчины с дополнительной Y-хромосомой обычно не имеют существенных отличий от мужчин с обычным набором хромосом, но могут иметь ряд особенностей. Клинические проявления: как и при трисомии-Х у женщин, определенного «синдрома», т. е. клинически специфической симптоматики, позволяющей диагностировать наличие добавочной Y-хромосомы без цитогенетического обследования, не имеется. Наиболее частым признаком является высокий рост, который у взрослых больных составляет в среднем 186 см. Однако этот признак не является абсолютным, так как в литературе имеются описания мужчин с кариотипом 47, XYY среднего роста. У части больных отмечаются нерезко выраженные евнухоидные черты телосложения и диспластические признаки: неправильное строение зубов, увеличение нижней челюсти, аномальный прикус, девиация коленных и локтевых суставов, радиоульнарный синостоз. У некоторых больных обнаруживается повышение уровня андрогенов и лютеинизирующего гормона. Половая функция не нарушена. Наличие добавочной Y-хромосомы может и не сопровождаться клинической патологией, но, несомненно, оно коррелирует как с интеллектуальным недоразвитием, так и с эмоционально-волевыми нарушениями. Не случайно наибольшая частота синдрома XYY обнаружена среди высокорослых преступников. В этой категории она составляет в разных исследованиях от 3 до 10 %. Синдром Патау (синдром трисомии 13 пары) •Кариотип 47 ХХ или ХУ, 13+. Среди больных преобладают девочки. •Дети рождаются обычно в срок, но с истинной пренатальной гипоплазией. Наблюдается высокая младенческая смертность (до 90% детей). Часть погибает внутриутробно. Клиника: микроцефалия; микрофтальм, анофтальмия; одно или двустороннее незаращение верхней губы и неба; полидактилия, выпуклые ногти, поперечная ладонная складка, повышенная гибкость суставов; множественные пороки развития нервной системы и внутренних органов - аплазия мозолистого тела, гипоплазия мозжечка, врожденные пороки сердца (дефект межжелудочковой перегородки, дефект меж предсердной перегородки), аномалии почек, пороки развития органов пищеварения (незавершенный поворот кишечника, дивертикул Меккеля); ушные раковины неправильной формы, низко расположены; крипторхизм, гипоплазия наружных половых органов, гипоспадия у мальчиков, удвоение матки и влагалища, двурогая матка у девочек; апноэ; судорожный синдром. Синдром кошачьего также болезнь кошачьего крика, синдром Лежёна — по имени описавшего его в 1963 году французского учёного) — редкое генетическое расстройство, вызываемое отсутствием фрагмента 5-й хромосомы. Кариотип 46 XX или XY, 5р-. Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом. Хромосомный синдром кошачьего крика объясняется частичной моносомией; он развивается при делеции (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а утрата конкретного короткого фрагмента хромосомы. Изредка отмечается мозаицизм по делеции или образование кольцевой хромосомы-5. При этом синдроме наблюдается: · общее отставание в развитии, · низкая масса при рождении и мышечная гипотония, · лунообразное лицо с широко расставленными глазами, ·характерный плач ребёнка, напоминающий кошачье мяуканье, причиной которого является изменение гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани. Признак исчезает к концу первого года жизни. Кроме того, встречаются врождённые пороки сердца, костно-мышечной системы и внутренних органов, микроцефалия, птоз, низкое расположение и деформация ушных раковин, кожные складки впереди уха, гипертелоризм (увеличенное расстояние между какими-либо парными органами или анатомическими образованиями — например, между внутренними краями глазниц, грудными сосками), эпикантус (поперечная кожная складка около внутреннего угла глаза, обычно двусторонняя; наиболее чётко выражена при синдроме Дауна), антимонголоидный разрез глаз. 12. Взаимодействие аллельных генов: полное и неполное доминирование, сверхдоминирование, кодоминирование. Множественные аллели. Наследование групп крови человека по системе АВО. Неполное доминирование наблюдается, когда фенотип гетерозигот BB' отличается от фенотипа гомозигот по обоим аллелям (BB или B'B') промежуточным проявлением признака. Это объясняется тем, что аллель, способный сформировать нормальный признак, находясь в двойной дозе у гомозиготы BB, проявляется сильнее, чем в единственной дозе у гетерозиготы BB'. Указанные генотипы отличаются экспрессивностью, т.е. степенью выраженности признака. Полное доминирование – это такой вид взаимодействия аллельных генов, при котором проявление одного из аллелей (А) не зависит от наличия в генотипе особи другого аллеля (А1) и гетерозиготы АА1 фенотипически не отличаются от гомозигот по данному аллелю (АА). В гетерозиготном генотипе АА1 аллель А является доминантным. Присутствие аллеля А1 никак фенотипически не проявляется, поэтому он выступает как рецессивный. Кодоминирование представляет собой такой тип взаимодействия аллельных генов, при котором каждый из аллелей проявляет свое действие. В результате этого формируется некий промежуточный вариант признака, новый по сравнению с вариантами, определяемыми каждым аллелем самостоятельно. Примером может служить формирование IV, или АВ-группы, крови у человека, гетерозиготного по аллелям IA и IB, которые по отдельности детерминируют образование II и III групп крови. Межаллельная комплементация относится к достаточно редко встречаемым способам взаимодействия аллельных генов. В этом случае возможно формирование нормального признака D у организма, гетерозиготного по двум мутантным аллелям гена D(D'D"). Сверхдоминирование — это явление преимущества класса гетерозигот по сравнению с возможными для данного гена и аллелей классами гомозигот. Множественный аллелизм — это существование в популяции более двух аллелей данного гена. В популяции оказываются не два аллельных гена, а несколько. Множественный аллелизм для генов, контролирующих системы несовместимости, выступает как фактор отбора, препятствующий образованию зигот и организмов определенных зигот. Примером множественного аллелизма является серия множественных аллелей s1, s2, s3, обеспечивающих самостерильность многих растений. Двенадцать различных состояний одного локуса у дрозофилы, обусловливающих разнообразие окраски глаз и т.д. Примером доминирования одного из аллелей в гетерозиготном генотипе может служить определение групповой принадлежности крови у человека по системе АВ0. Генотипы, содержащие аллель IA либо в гомозиготном состоянии, либо в сочетании с аллелем I0 (IAIA или IAI0), определяют развитие у человека второй группы крови (группа крови А). Такая же ситуация наблюдается и в отношении аллеля IB, обусловливающего формирование третьей, или В-группы крови. Следовательно, аллели IA и IB выступают как доминантные по отношению к аллелю I0, формирующему в гомозиготном состоянии I0I0 первую, или 0-группу крови. 21. Комбинативная изменчивость, её механизмы и биологическое значение: Комбинативная изменчивость связана с получением новых сочетаний генов в генотипе. Достигается это в результате 3 процессов: · Независимого расхождения хромосом при мейозе; · Случайного их сочетания при оплодотворении; · Рекомбинации генов благодаря кроссинговеру; Сами наследственные факторы (гены) при этом не изменяются, но новые сочетания их между собой приводят к появлению организмов с новым фенотипом. К комбинативной изменчивости примыкает явление гетерозиса. Гетерозис или «гибридная сила», может наблюдаться в 1ом поколении при гибридизации между представителями различных видов или сортов. Проявляется он повышением жизнеспособности, увеличением роста и др. особенностями. Механизмы комбинативной изменчивости: · Постоянные: случайное и независимое расхождение хромосом в анафазе 1 мейоза 1; случайная встреча гамет при оплодотворении; случайный подбор родительских пар · Непостоянные: кроссинговер, поведение МГЭ (мобильных генетических элементов). Значение комбинативной изменчивости: 1. возникает огромное гено- и фенотипическое разнообразие особей. 2. Повышаются адаптивные возможности. 3. Может возникнуть комбинация генов, которая проявится в фенотипе как болезнь, или исключит ее проявление. Мейоз и оплодотворение обеспечивают получение организмами нового поколения сбалансированного по дозам генов наследственного материала, на основе которого осуществляется развитие организма и отдельных его клеток. Благодаря этим двум механизмам в ряду поколений особей данного вида формируются определенные видовые характеристики и вид как реальная единица живой природы существует продолжительное время. Так как в воспроизведении потомства принимают участие 2особи, то в результате оплодотворения зиготы получают неодинаковый набор аллелей в их генотипах. Увеличению генотипического разнообразия представителей вида способствуют также механизмы, приводящие к перекомбинации родительских аллелей особи в ее гаметах. Механизмом, обеспечивающим разнообразие гамет, является мейоз, в ходе которого происходит не только уменьшение вдвое наследственного материала, но и эффективное перераспределение родительских аллелей между гаметами. Процессами, приводящими к перекомбинации генов и целых хромосом в половых клетках, являются кроссинговер и расхождение бивалентов в анафазе I мейоза. Геном как высший уровень организации наследственного материала благодаря мейозу и оплодотворению сохраняет свои видовые характеристики. Но одновременно эти же процессы обеспечивают индивидуальные наследственные различия особей, в основе которых лежит рекомбинация генов и хромосом, т.е. комбинативную изменчивость. Комбинативная изменчивость, проявляющаяся в генотипическом разнообразии особей, повышает выживаемость вида в изменяющихся условиях его существования. 22. Понятие о мутационной изменчивости. Мутации в зависимости от места возникновения (соматические, генеративные), их значение. Примеры. Основные положения мутационной теории: Мутация – изменение, обусловленное реорганизацией воспроизводящих структур, изменением ее генетического аппарата. Мутации возникают внезапно, скачкообразно, что иногда резко отличает организм от исходной формы. С мутационной изменчивостью связана эволюция – процесс образования новых видов, сортов и пород. По характеру изменения генетического аппарата различают мутации: · Изменением числа хромосом (геномные); · Изменением структуры хромосом ( хромосомные аберрации); · Изменением молекулярной структуры гена ( генные, или точковые мутации). Любые мутационные изменения в наследственном материале гамет —генеративные мутации —становятся достоянием следующего поколения, если такие гаметы участвуют в оплодотворении. Поэтому отклонения в течении митоза или мейоза в клетках-предшественницах гамет имеют большое эволюционное значение. Если же мутации любого ранга (генные, хромосомные или геномные) возникают в соматических клетках — соматические мутации — они передаются только потомкам этих клеток, т.е. не выходят за пределы данного организма. Исключение составляют соматические мутации, возникшие в клетках органов вегетативного размножения, от которых они передаются новому поколению организмов. Одной из причин соматических мутаций являются патологические митозы. При нарушении нормального течения митоза развитие дочерних клеток отклоняется от нормы. Патологические митозы часто наблюдаются в клетках злокачественных опухолей. Таким образом, несмотря на существование механизмов, обеспечивающих стабильность структуры генома, на этом уровне организации наследственного материала могут появляться эволюционно значимые изменения. Они способны обеспечить достаточно резкий скачок в ходе исторического развития живой природы. Основные положения мутационной теории Коржинского — Де Фриза: 1. Мутации внезапны, как дискретные изменения признаков. 2. Новые формы устойчивы. 3. В отличие от ненаследственных изменений, мутации не образуют непрерывных рядов, не группируются вокруг какого-либо среднего типа. Они являют собой качественные скачки изменений. 4. Мутации проявляются по-разному и могут быть как полезными, так и вредными. 5. Вероятность обнаружения мутаций зависит от числа исследуемых особей. 6. Сходные мутации могут возникать неоднократно. 30. Сущность молекулярных болезней человека. Возможности их профилактики и лечения. Серповидноклеточная анемия, фенилкетонурия, болезнь Вильсона-Коновалова, муковисцидоз, наследственная гиперхолестерененимия, идиотия Тея-Сакса. Молекулярные болезни – дефект на молекулярном уровне, нарушение в структуре молекулы ДНК. В зависимости от то мутации, ферментативная активность может быть изменена: повышена, понижена, отсутствует. Такие мутации проявляются как наследственные болезни обмена веществ – ферментопатии (энзимопатия), вещества, накапливающиеся в результате выпадения ферментов, продукты их реакция, оказывают побочное действие на организм (болезнь накопления). Заболевания, связанные с генной мутацией могут, проявляются как с момента рождения, так и после, даже в старости. Расстройства липидного обмена могут сопровождаться увеличением концентрации липидов в сыворотке крови, понижение концентрации липидов или отложением этих веществ в клетках тканей, где в норме этого нет, например такое накопление может происходить в нервных клетках, печени, почках при болезни Тая-Сакса. (ИНТЕРНЕТ: нейродегенерация, «Вишневая косточка» - на области сетчатки розовое пятнышко, судороги, паралич, психоз, теряет зрение и слух). Рецессивный тип. Нарушение обмена меди, которая входит в состав ферментов, участвующие в реакциях окисления. При определенной генной мутации тормозится синтез белка церрулоплазмина, поэтому в крови уменьшается количество меди и она накапливается в тканях печени и мозга, вызывая дегенерацию – болезнь Вильсона-Коновалова. Лечат уменьшением поступления меди или ее выведением. С 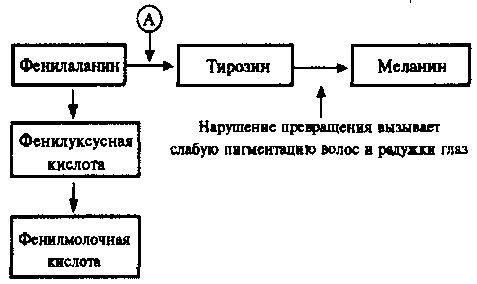 ерповидно-клеточная анемия заключается в замене гемоглобина HbA и HbS, который отличается растворимостью и кристаллизацией в условиях гипоксии, что приводит к изменению формы эритроцитов и проявляется фенотипическим многообразием симптомов. Наследуется по рецессивному типу, но и в условиях сильной гипоксии (гетерозиготы при нахождении свыше 3000м над уровнем моря также страдают анемии). ерповидно-клеточная анемия заключается в замене гемоглобина HbA и HbS, который отличается растворимостью и кристаллизацией в условиях гипоксии, что приводит к изменению формы эритроцитов и проявляется фенотипическим многообразием симптомов. Наследуется по рецессивному типу, но и в условиях сильной гипоксии (гетерозиготы при нахождении свыше 3000м над уровнем моря также страдают анемии). Фенилкетонурия – нарушение процессов обмена аминокислоты фенилалланин и накопления в организме токсических промежуточных продуктов. При дефекте фермента фенилалланингидроксилазы финилалланина не превращается в тирозин, это приводит к превращению фенилалланина в фенилоуксуную к-ту и фенилмолочную к-ту = токсичное действие на мозг ребенка=дефект умственного развития = также нарушение синтеза мелатонина= слабая пигментация волос и радужки глаз. Наблюдается судорожный синдром, нарастает отставание развития. Наследуется рецессивно. Муковисцидозы — встречаются с частотой 1:2500 новорожденны наследуются по аутосомно-рецессивному типу. В основе патогенеза заболевания — наследственное поражение экзокринных желез (поджелудочной, желез кишечника, дыхательных) и железистых клеток организма, выделение ими густого, измененного по составу секрета и связанные с этим последствия, вязкий секрет закупоривает выводные протоки желёз, развивается воспалительный процесс с присоединением вторичной инфекции (Увеличенная концентрация гидрокарбоната и хлорида натрия в поте., одышка, кашель, повторные бронхиты и пневмонии, синуситы, экзокринная недостаточность поджелудочной железы: вздутие живота, частый, обильный, зловонный маслянистый стул) Гиперхолестеринемия — это те нарушения липидного состава крови, которые сопровождаются повышением в ней концентрации холестерина. Из возможных проявлений резко выраженной гиперхолестеринемии можно назвать: атеросклеротические бляшки в сосудах; ксантелазмы — желтоватые слегка выступающие над кожей образования на веках; ксантомы — жёлтые или оранжевые отложения липидов в коже или сухожилиях; липоидную дугу роговицы = белёсую дугу или ободок вокруг радужки глаза (лечить: уменьшение потребления таких продуктов, как жирное мясо, жирные молочные продукты, кондитерские изделия и сладости; полное прекращение курения; снижение массы тела хотя бы на 10% от исходного, если имеется избыточная масса тела и ожирение). 31. Наследственные болезни с нетрадиционным наследованием (митохондриальные болезни: пример с наследованием зрительной невропатии Либера) Митохондриальные болезни. В 1980-х годах получены доказательства связи наследственных болезней с мутациями мтДНК. Выделют такие типы мутаций митохондриальных болезней: а) точковыми мутациями - замена ак-т в собственных белках митохондрий = пигментный ретинит и нейроофтальмопатия Лебера, при которой наступает двусторонняя потеря зрения. Выраженность клинических признаков зависит от количества мутантной мтДНК, которое может варьировать от 5 до 100% всей мтДНК; б) мутаций в генах т-РНК приврдящие к многим дегенеративным заболеваниям с различной степенью тяжести клинич. проявлений, коррелирующей с количеством мутантной мтДНК; в) болезни вызванные делениями и дупликациями участков митохондриалъных генов. У человека описано тяжелое заболевание молодого и среднего возраста — отсроченная кардиопатия – происходит делеция мтДНК кардиоцитов (Х-сцепленное наследование), считают что существует ряд генов из-за которого происходит деление до 50% мтДНК кардиоцитов; г) снижением числа копий мтДНК = мутаций. К данной группе относятся летальная инфантильная дыхательная недостаточность и синдром молочнокислого ацидоза, где число копий мтДНК снижается до 1—2% от нормы – это приводит к развитию миопатий, нефропатий, печеночной недостаточности и т.д. Изменения мтДНК = нарушению их функций, связанных с клеточным дыханием = это опеределяет характер и степень тяжести клинических проявлений митохондриалъных болезней. Выдвинута также гипотеза о том, что накопление спонтанных мутаций мтДНК является звеном механизмов старения и развития дегенеративных процессов у человека. ПРИМЕР ИЗ ИНЕТА: Наследственная оптическая нейропатия Лебера явл. наследственной митохондриальной дегенирацией ганглионарных клеток сетчатки и их аксонов, что приводит к острой потере зрения (сцеплено с Х-хромосомой рецессивно; от матери к детям; преимущественно влияет на мужчин). Еще называют «громом среди ясного неба» из-за изображения на сетчатке. Было впервые описано немецким офтальмологом Теодором Лебером в 1871 году, описал четыре семьи в которых молодые люди страдали от резкой потери зрения в обоих глазах одновременно или последовательно. Клиника: острое начало потери зрения, сначала в одном глазу, а затем через промежуток времени от нескольких недель до нескольких месяцев - в другом. Начинается обычно в юности, но может быть в диапазоне 7-75 лет. Прогноз – неизлечимая болезнь, продолжение снижения зрения в обоих глазах. Как правило рекомендуется избегать цианокобаламин (витамин В12), табак и алкоголь. Некоторые отпускаемые по рецепту лекарства, как известно, несут потенциальный риск, так что ко всем препаратам следует относиться употреблять с осторожностью. 34. Основные направления генной инженерии и терапии. Генная терапия моногенных болезней. Генная инженерия - совокупность методов молекулярной генетики, направленных на искусственное создание новых, не встречающихся в природе сочетаний генов, преобразование определенных функций организма, при внесении заданных свойств или коррекции генетических аномалий. Направления: 1)Генная селекция растений, животных и бактерий с целью повышения продуктивности, устойчивости к болезням и абиотическим факторам и внедрения генов животных в гены растений. 2)Производство источников энергии и новых материалов: бензин заменяют этиловым спиртом, полученный бактериями из растительного сырья. Использование «биогаза», искусственной нефти, солярки из бытовых отходов. Производство искусственных тканей с помощью микроорганизмов. 3) Генная инженерия в медицине: производство лекарств (инсулин, интерферон, соматотропин, антибиотики, вакцины, витамины), генная терапия: выделение поврежденного гена и переноса нормального в клетку (генные болезни обмена веществ). Генная терапия — совокупность генноинженерных и медицинских методов, направленных на внесение изменений в генетический аппарат соматических клеток человека в целях лечения заболеваний, вызванных мутациями (изменениями) в структуре ДНК, или придания клеткам новых функций, например, подавление излишней функции генов, также, исправление ошибок — искусственном создании необходимых количеств продуктов того или иного плохо работающего гена в организме больного, за счет введения в организм нормального гена, полученного от здоровых пациентов. Для моногенных заболеваний, вызванных мутациями с потерей функции гена, лечение направлено на замену дефектного белка, улучшение его функции или минимизацию последствий недостаточности. Замену дефектного белка можно достичь его введением, пересадкой органа или клеток, или генотерапией. ПРИМЕРЫ: Муковисцидоз - Показано, что замена 6-10% клеток легочного эпителия трансфецированными клетками позволит восстановить нормальные транспортные функции трансмембранных каналов, обеспечивающих перенос ионов хлора. Решение данной проблемы заключается в модификации вектора, который включает в себя определенный лиганд к рецептору на поверхности клеток легочного эпителия. Лиганда + рецептор = приводит к переходу вектора вместе с рецептором внутрь клетки. В качестве такого рецептора был выбран трансмембранный рецептор P2Y2-R - участвует в запуске каскада воспалительных реакций в полости легких. В качестве лиганда использовались либо моноклональные антитела к этому рецептору, либо природный лиганд - биотинУТФ. Недостаточность аденозиндезаминазы(АДА синдром) – первый опыт в 1990г - с помощью лейкофореза из периферической крови выделяли мононуклеарные клетки, затем их растили, после в условиях in vitro клетки вводили ретровирусный вектор, который содержал нормальный ген ADA. Через несколько дней эти клетки крови вводили обратно пациентке. Процесс повторяли 7 раз на протяжении 10 месяцев. Эффект был положительным, ¼ лифоцитов в организме получили рабочий ген. |