Функции печени
 Скачать 1.43 Mb. Скачать 1.43 Mb.
|

|
Функции печени Депо гликогена, крови, железа, витаминов В12 и жирорастворимых. Выработка желчи, участие в пищеварении. Детоксикационная «барьерная» функция, в отношении веществ поступающих из кишечника. Участие в белковом обмене: альбумины 100%, свертывающая и противосвертывающая системы, переаминирование и дезаминирование аминокислот, образование мочевины из аммиака (инактивация аммиака). Участие в углеводном обмене: глюконеогенез, гликогенез, гликогенолиз, синтез глюкуроновой кислоты. Участие в жировом обмене: синтез триглицеридов, фосфолипидов, холестерина, разрушение избытка жирных кислот. Участие в пигментном обмене: выведение продуктов распада эритроцитов. Участие в обмене витаминов: всасывание жирорастворимых (ч/з желчь), депонирование (в т.ч. В12), образование витамина А из каротина, разрушение «отслуживших» витаминов. Участие в обмене микроэлементов: железа, меди, кадмия и других. Участие в обмене ферментов: образование основных или готовых ферментов. Участие в обмене гормонов: разрушение и инактивация гормонов, в т.ч. эстрогенов, альдостерона, гастрина, с другой стороны синтез тирозина – предшественника адреналина и норадреналина, Т3,4. Участие в обмене БАВ, влияющих на тонус сосудов, например, производных арахидоновой кислоты, кининов и др., вызывая дилатацию сосудов большого круга, на фоне спазма прекапиллярных сосудов малого круга кровообращения. Участие в терморегуляции через пирогенный стероид этиохоланолон, который печень инактивирует. Орган кроветворения у плода. Обмен желчных пигментов Основной желчный пигмент — билирубин — представляет собой конечный продукт обмена гема. Главным источником билирубина крови является гемоглобин эритроцитов. Образование билирубина из гема происходит в клетках РЭС. Последовательность биохимических превращений следующая: гемоглобин → вердоглобин → биливердин → билирубин. Образовавшийся билирубин не растворим в воде, поэтому его транспорт в печень осуществляется в связанном с белками виде (неконъюгированный). С током крови он попадает в печень, где происходит три процесса, важных с точки зрения обмена желчных пигментов: захват гепатоцитами связанного с белками (непрямого) билирубина - обеспечивается специфическими белковыми рецепторами плазматической мембраны печеночных клеток; конъюгация билирубина с глюкуроновой кислотой, вследствие чего образуются глюкурониды билирубина (конъюгированный билирубин); экскреция прямого билирубина в составе желчи. Все 3 процесса требуют затрат АТФ. Образовавшийся конъюгированный билирубин попадает в желчный пузырь, а от туда в тонкую кишку, где под действием ферментов микрофлоры превращается в уробилиноген. Основная масса уробилиногена в толстой кишке преобразуется в стеркобилиноген, который в прямой кишке превращается в стеркобилин под влиянием воздуха и выводится с калом. Часть стеркобилиногена всасываясь в кровь в области нижнего и среднего геморроидальных сплетений прямой кишки, экскретируясь почками, придавая моче желтоватый оттенок. Вторая, намного меньшая часть уробилиногена принимает участие в так называемом печеночно-кишечном кругообороте — всасывается в тонкой кишке, попадает в печень, частично подвергается окислению до ди и трипиролов, а частично вновь поступает в желчевыводящие пути и кишечник. NB!Из обращения следует исключить использование связанный и несвязанный билирубин, как устаревшие термины, в настоящее время правильно использовать неконъюгированный (образованный при гемолизе, неконъюгированный ЕЩЁ с глюкуроновой кислотой) и конъюгированный (связанный с глюкуроновой кислотой – более токсичный чем неконъюгированный). Желтухи Желтуха (icterus) — это синдром, обусловленный увеличением уровня билирубина в крови и проявляющийся желтым окрашиванием кожи и слизистых оболочек. Выделяют три вида желтухи: гемолитическая (надпеченочная) желтуха, паренхиматозная (печеночная) желтуха, механическая (обтурационная, или подпеченочная) желтуха. Гемолитическая (надпеченочная) желтухаГемолитическая желтуха развивается вследствие ускоренного гемолиза эритроцитов. Усиленный фагоцитоз эритроцитов или самого гемоглобина, высвободившегося из разрушенных эритроцитов, приводит к образованию в фагоцитах больших количеств неконъюгированного билирубина, который затем поступает печень. Гепатоциты при этом испытывают повышенную нагрузку, преобразуя большие количества непрямого билирубина в конъюгированный и экскретируя последний в составе желчи. Этим объясняется высокое содержание стеркобилина в кале (гиперхоличный кал) и стеркобилиноегна моче. В связи с тем, что неконъюгированный билирубин не фильтруется в почках (связан с белками), его в моче нет. Итак основные признаки гемолитической желтухи: а) увеличение содержания общего билирубина в крови, за счёт фракции неконъюгированного билирубина; б) увеличение содержания стеркобилина в кале (гиперхоличный кал); в) увеличение содержания стеркобилиногена в моче; Паренхиматозные (печеночные) желтухиВ зависимости от патогенеза выделяют печеночно-клеточную желтуху, желтуху из-за нарушения процесса захвата билирубина, желтуха из-за расстройства процесса конъюгации и желтуха из-за нарушения процесса экскреции билирубина. В клинической практике чаще встречается печено-клеточная желтуха. Печеночно-клеточная желтуха Характеризуется нарушениями всех трех процессов, происходящих в гепатоцитах: захвата, конъюгации и экскреции билирубина. Возникает при повреждении гепатоцитов и сопровождает печеночно-клеточную недостаточность. Вследствие гибели печеночных клеток уменьшается масса работающих гепатоцитов, что приводит к нарушению захвата и дальнейшей метаболической обработки («работников» становится мало). Сохранившиеся гепатоциты находятся, ка правило, под влиянием этиологического фактора повреждающего клетки, т.е. даже сохранившиеся гепатоциты не в состоянии качественно и быстро метаболизировать еще неконъюгированный билирубин, т.к. АТФ требуется на это много (малочисленные оставшиеся «работники» больны). При повреждении гепатоцитов закономерно увеличивается проницаемость гепатоцитов, дополнительно если гепатоциты погибнут – нарушается целостность желчного капилляра - образуются сообщения между желчными и кровеносными капиллярами. В первом и во втором случае желчь попадает в кровь (холемия), а вместе с ней и конъюгированный билирубин с желчными кислотами. А из-за уменьшения способности печени конъюгировать и выделять в желчевыводящие пути билирубин («работников» мало, а оставшиеся больны) уменьшается поступление желчи в кишки (гипохолия), что приводит к диспепсии и снижению интенсивности окраски кала. В связи с указанными нарушениями появляются следующие изменения показателей пигментного обмена: а) увеличение содержания в крови общего билирубина за счет фракций как конъюгированного, таки неконъюгированного билирубина; в) уменьшение содержания стеркобилиногена в кале (гипохолич- ный кал) – окрашен слабо, но всё же окрашен, т.к. какое-то количество желчи выделяется; г) появление в моче билирубина, т.к. конъгированный билирубин попадает в кровь из-за повреждения и разрушения гепатоцитов, конъюгированный билирубин водорастворим и проходит через почечный фильтр; д) появление в моче уробилиноидов, т.к. всосавшись в тонкой кишке они должны били попасть по портальной системе в печень и там связаться гепатоцитами, разрушится до ди и трипиролов и выделиться с желчью, однако гепатоцитов меньше, а оставшиеся функционируют плохо, в результате уробилинойды «проскакивают» через печень, попадают в кровоток и выделяются почками ; д) уменьшение или полное отсутствие стеркобилиногена в моче. Кроме того, в крови и моче обнаруживаются желчные кислоты (холалемия и холалурия). Печеночные желтухи с изолированными нарушениями процессов, обеспечивающих выведение билирубина из организма. Нарушения захвата непрямого билирубина (синдром Жильбера — наследственно обусловленный дефицит рецепторов к белок-билирубиновому комплексу). Проявляется увеличением содержания непрямого билирубина в крови и уменьшением количества стеркобилиногена в кале и моче; в целом благоприятное течение (без фиброза и цирроза), однако в последнее время появились данные свидетельствующие о повышении риска желчекаменной болезни. Расстройства конъюгации билирубина (синдром Криглера-Наджара). Они чаще всего связаны с приобретенным или наследственным дефицитом фермента глюкуронилтрансферазы. Проявляются увеличением содержания непрямого билирубина в крови и уменьшением содержания стеркобилиногена в кале и моче. Нарушения экскреции билирубина (синдром Дабина-Джонсона, синдром Ротора). Их причиной являются дефекты (чаще всего наследственные) систем транспорта билирубина и желчи из гепатоцитов в желчные капилляры. Проявляются увеличением содержания прямого билирубина в крови, появлением в крови и моче желчных кислот, билирубинурией, уменьшением содержания или полным отсутствием стеркобилиногена в кале и моче. Механическая (подпеченочная) желтухаРазвивается в результате механического препятствия оттоку желчи. Это может быть следствием: 1) сдавливания желчевыводящих путей извне (опухоль головки поджелудочной железы, действие рубца); 2) их закупорка камнем, гельминтами, густой желчью. Механическое препятствие оттоку желчи приводит к застою и повышению давления желчи, расширению и разрыву желчных капилляров и поступлению желчи прямо в кровь или через лимфатические пути. Возникают следующие изменения пигментного обмена: а) увеличивается содержание в крови общего билирубина за счёт фракции конъюгированного билирубина (гипербилирубинемия); б) в крови появляются желчные кислоты (холалемия); в) в моче появляется конъюгированный билирубин (билирубинурия), желчные кислоты (холалурия), вследствие чего она приобретает темную окраску («цвет пива»), но из мочи исчезает стеркобилиноген; г) в кале не содержится стеркобилин (бесцветный кал). Перечисленные изменения пигментного обмена обусловливают развитие двух важных клинических синдромов, характерных для механической желтухи: холемического и ахолического. Холемический синдром (синдром холестаза) обусловлен поступлением компонентов желчи (желчных кислот, конъюгированного билирубина, холестерина) в кровь в связи с нарушением формирования и оттока желчи. Он закономерно возникает при механической желтухе, а также некоторых формах печеночной желтухи (печеночно-клеточной, печеночной желтухе, обусловленной нарушениями экскреции желчи). Основные проявления синдрома обусловлены: 1. Появлением в крови желчных кислот — холалемии: расстройства деятельности центральной нервной системы, возникающие как следствие общетоксическогодействияжелчных кислот (общая астения, раздражительность, сменяющаяся депрессией; сонливость днем и бессонница ночью; головные боли, утомляемость); артериальная гипотензия, брадикардия. Их развитие обусловлено повышением тонуса блуждающего нерва и непосредственным действием желчных кислот на синусовый узел и кровеносные сосуды; зуд кожи, возникающий в результате раздражения нервных окончаний желчными кислотами; множественные повреждения и гибель клеток, обусловленные детергентным действием желчных кислот. Этим, в частности, объясняется гемолиз эритроцитов, воспаления и некрозы в разных органах и тканях; появление желчных кислот в моче (холалурия). 2. Поступлением в кровь конъюгированного билирубина. Это обстоятельство вызывает появление желтого окрашивания кожи и слизистых оболочек, т.е. собственно желтухи с зеленоватым оттенком. 3. Увеличением содержания в крови холестерина и появление аномального липопротеида X, обладающего высоко атерогенным действием. Ахолическим называют синдром, обусловленный отсутствием желчи в кишечнике в связи с нарушениями ее формирования и оттока. Характерны: 1. Расстройства переваривания и всасывания жиров. Обусловлены нарушением процессов эмульгирования жиров, уменьшением активности панкреатической липазы, активируемой желчью. Следствием указанных изменений являются: появление жира в кале — стеаторея; расстройства всасывания жирорастворимых витаминов, в результате чего развиваются гиповитаминозы D,E,K,A; уменьшение поступления в организм ненасыщенных жирных кислот, необходимых для построения фосфолипидов клеточных мембран. 2. Нарушения двигательной функции кишечника и усиление гнилостных процессов и реакций брожения в результате уменьшения бактерицидного действия желчи. Это приводит к увеличению нагрузки на антитоксические системы печени. 4. Обесцвечивание кала. Дисхолия, механизм возникновения желчных камнейДисхолия — это нарушения физико-химических свойств желчи, вследствие чего она приобретает литогенные свойства, т.е. способность образовывать камни (конкременты) в желчном пузыре и желчных протоках. Результатом этого является развитие желчнокаменной болезни. Факторы риска желчнокаменной болезни: женский пол; зрелый возраст; избыточная масса тела; несколько беременностей в анамнезе; лечение клофибратом и эстрогенами; нерациональное питание; инфекционно-воспалительные процессы в желчном пузыре и желчных протоках; застой желчи (холестаз). Одним из основных механизмов возникновения литогенной желчи является снижение отношения желчных кислот и лецитина к холестерину желчи. Это может быть вызвано уменьшением печеночно-кишечного кругооборота желчных кислот при патологии кишечника и изменении его микрофлоры, угнетением синтеза желчных кислот в печени, ускорением их всасывания слизистой оболочкой воспаленного желчного пузыря, уменьшением содержания лецитина и увеличением синтеза холестерина. При уменьшении концентрации желчных кислот и лецитина, обеспечивающих взвешенное состояние холестерина, холестерин выпадает в осадок и дает начало образованию холестериновых камней. Инфекция, застой желчи также способствуют процессу камнеобразования, так как сопровождаются изменением свойств желчи — сдвигом рН в кислую сторону, снижением растворимости солей, выпадением их в осадок, коагуляцией белков из распадающихся клеток. Есть и другие литогенный факторы, например, кишечная палочка, синтезируют фермент Р-глюкуронидазу, которая превращает растворимый конъюгированный билирубин в неконъюгированный, выпадающий в осадок. В целом по аналогии с камнеобразованием в почках можно выделить две главные теория развития камней: кристаллизационная теория и теория белковой матрицы. Кристаллизационная теория указывает на то, что вначале происходит концентрирование желчи, на этом фоне образуется маленький кристалл-затравка, вокруг которого начинается кристаллизация и каменеобразование. Этот механизм похож на технологию выращивания неорганических кристаллов. Теория белковой матрицы объясняет, что на фоне воспаления в желчном пузыре образуется белковая матрица (фибрин, слущенная клетка эпителия или др.) на них начинается процесс кристаллизации. Так или иначе, процесс формирования желчных камней включает в себя три стадии: насыщение, кристаллизацию и рост. Помимо холестериновых, образуются пигментные (при гемолизе эритроцитов), кальциевые и сложные камни (например, холестериново-пигментно-кальциевые). Камни обусловливают нарушение желчевыделения и развитие механической желтухи. Цирроз печениОпределение Это хроническое полиэтиологическое диффузное прогрессирующее заболевание, характеризующееся появлением узлов регенерации, значительным уменьшением количества функционирующих гепатоцитов, нарастающим фиброзом, перестройкой нормальной структуры паренхимы и сосудистой системы печени, развитием в последующем печеночной недостаточности и портальной гипертензии. |NB!Ростислав Владимирович делает БОЛЬШОЙ акцент на то, что именно узлы регенераты – главный морфологический признак цирроза, отражающий суть его патогенеза. Т.к., фиброз, преходящая портальная гипертензия могут быть и при гепатите с высокой степенью активности воспаления. Этиология Стартовым механизмом цирроза является некроз гепатоцитов и длительно текущее в печени воспаление. Ниже перечислены этиологические факторы, которые могут вызывать массивный некроз и/или длительно поддерживающееся воспаление: вирусные гепатиты - имеют значение персистирование вирусной инфекции и обусловленного ею иммуновоспалительного процесса, цитопатическое (гепатотоксическое) действие вирусов D и С, развитие аутоиммунных реакций; алкоголь и другие токсические вещества (удобрения, ароматические углеводороды, токсины грибов и бактерий) - ведущее значение приобретают повреждение гепатоцитов алкоголем и продуктом его метаболизма - ацетальдегидом, развитие аутоиммунного воспалительного процесса (в ответ на отложение в печени алкогольного гиалина), стимуляция фиброзирования в печени под влиянием алкоголя; аутоиммунные процессы - основную роль играют аутоиммунные реакции, вызывающие резко выраженный иммуновоспалительный процесс с некрозами печеночной ткани; наследственные болезни (недостаточность α1-антитрипсина, болезнь Уилсона–Коновалова, гемохроматоз); некоторые лекарственные препараты (противотуберкулезные препараты, цитостатики, стероидные гормоны, метилдофа); хронические болезни желчевыводящих путей; паразитарные инвазии (шистосомоз, клонорхоз). В ряде случае (до 20% всех циррозов) бывает Ваш любимый - идиопатический цирроз. Патогенез цирроза печени Главным в патогенезе является запущенная некрозом и длительным воспалением активная пролиферация гепатоцитов, но без сохранения нормальной гистоструктуры долек, а также развитие фиброза и тяжелых расстройств регионарной гемодинамики внутри печени. Клиническим результатом патогенетических путей цирроза становятся гепатоцеллюлярная недостаточность и портальная гипертензия. Рассмотрим ниже частные вопросы патогенеза цирроза печени. Патогенез цирроза печени: гибель гепатоцитов и образование узлов Итак, пусковым фактором в патогенезе циррозов является гибель печеночной паренхимы и спадение ретикулярного остова на месте погибших гепатоцитов. В нормальных условиях портальная вена и печеночная артерия отдают свою кровь в синусоиды, расположенные между балками гепатоцитов в дольке, а затем кровь попадает из синусоидов в центральную (печеночную) вену. Однако в результате некроза гепатоцитов и спадения ретикулярного остова сосуды портального тракта приближаются к центральной вене. Создаются условия для перехода (сброса) крови из печеночной артерии и воротной вены в центральную вену, минуя синусоиды расположенных рядом неповрежденных участков печени. Ток крови в обход синусоидов неповрежденных участков печени приводит к их ишемии, а затем и некрозу. При первичном некрозе гепатоцитов (под влиянием этиологического фактора) и вторичном некрозе (в результате ишемии) выделяются стимулирующие регенерацию печени вещества, однако регенерация осуществляется без сохранения нормальной гистоархитектоники дольки, а с образованием узла-регенерата (давайте между собой называть его узлом-дегенератом, т.к. в нем абсолютный хаос в плане расположения гепатоцитов, сосудов, но об этом чуть ниже). В патогенезе цирроза печени, а именно в неадекватной пролиферации, играют значение кейлоны. Их количество уменьшается из-за уменьшения массы паренхимы печени, что приводит к высокому пролиферативному потенциалу в оставшихся гепатоцитах, которые делятся, однако не пытаются сохранить нормальную гистоархитектонику. Образовавшиеся узлы регенераты (дегенераты), сдавливают сосуды и способствуют дальнейшему нарушению кровотока в печени. Патогенез цирроза печени: гибель гепатоцитов и воспаление Важно, что продукты распада гепатоцитов стимулируют воспалительную реакцию, формируются воспалительные инфильтраты, которые распространяются из портальных полей до центральных отделов долек и сдавливают синусоиды и центральную вену дольки (синусоидальные и постсинусоидальный блок). Вы помните, что любой воспалительный процесс заканчивается процессом регенерации клеточного состава и соединительно-тканного каркаса. Однако при циррозе аномально проходит не только регенерация гепатоцитов с образованием узлов регенератов (дегенератов), аномально проходит и восстановление соединительно-тканного каркаса с интенсивным избыточным фиброзообразованием (подробнее об этом смотри ниже в разделе «Патогенез цирроза печени: фиброз»). Патогенез цирроза печени: фиброз Главные участники фиброгенеза: клетки Купфера (макрофаги печени), которые активно продуцируют профибротические цитокины; клетки Ито (звездчатые клетки), расположены в перисинусоидальном пространстве (Диссе), при циррозепечени трансформируются в мифибробласты, продуцирующие коллаген. При развитии фиброза увеличивается количество синтезируемого коллагена I и III типов более чем в 3-10 раз, перераспределяется соединительная ткань в пространство Диссе. В развитии фиброза выделяют 3 фазы: инициации, пролонгации и резолюции. Фаза инициации – суть заключается в активации клеток Купфера, которые будут продуцировать провоспалительные и профибротические цитокины. Механизмы активации клеток Купфера следующие: поврежденные гепатоциты выделяют активные формы кислорода (вспомните механизмы повреждения клетки), что активирует клетки Купфера; активация клеток Купфера из-за альтерации эндотелия синусоидов этиологическим фактором или продуктами повреждения гепатоцитов (лизосомальные ферменты, продукты СПОЛ, фосфолипазы, производные арахидоновой кислоты); Фаза пролонгации. В предыдущей фазе активизировались клетки Купфера, они выделяют профибротические и провоспалительные цитокины, самые главные из них трансформирующий фактор роста β1(TGFβ1), тромбоцитарный фактор роста. Трансформирующий фактор роста и другие цитокины активируют следующего участника фиброза – клетки Ито (звездчатые клетки) в перисинусоидальном пространстве. Под влиянием цитокинов клетки Ито начинают активно мигрировать, пролиферировать и трансформироваться в миофибробласты. Миофибробласты начинают продуцировать коллаген I и III типов – так формируется фиброз и склероз. Кроме того миофибробласты начинают продуцировать протеогликаны, кислые гликозаминогликаны, эластин. По самым последним данным в процесс фиброза значительный вклад осуществляют мигрировавшие из костного мозга фибробласты и моноциты, трансформированные в фибробласты под влиянием TGFβ1. Резолюции – заключается в резорбции образовавшегося коллагена под влиянием металлопротеиназ, однако при циррозе этот процесс значительно подавлен. Увеличение количества соединительной ткани в перисинусоидальном пространстве приводит к тому, что закрываются фенестры и синусы превращаются в обычные капилляры – капилляризация синусоидов. Это нарушает нормальный обмен между кровью и гепатоцитами, приводит к гибели гепатоцитов. Разрастание соединительной ткани приводит также к формированию соединительнотканных септ. Они содержат сосудистые анастомозы, соединяют центральные вены и портальные тракты, а долька, мало того еще фрагментируется на псевдодольки. В псевдодольках полностью изменено взаимоотношение портальных сосудов и центральной вены, в центре псевдодолек не обнаруживается центральной вены, а по периферии нет портальных триад. Псевдодольки окружены соединительнотканными септами, содержащими сосуды, соединяющие центральные вены с ветвями печеночной вены (внутрипеченочные порто-кавальные шунты). Кровь поступает сразу в систему печеночной вены, минуя паренхиму псевдодолек, это вызывает ишемию и некроз. Этому также способствует механическое сдавление венозных сосудов печени соединительной тканью. 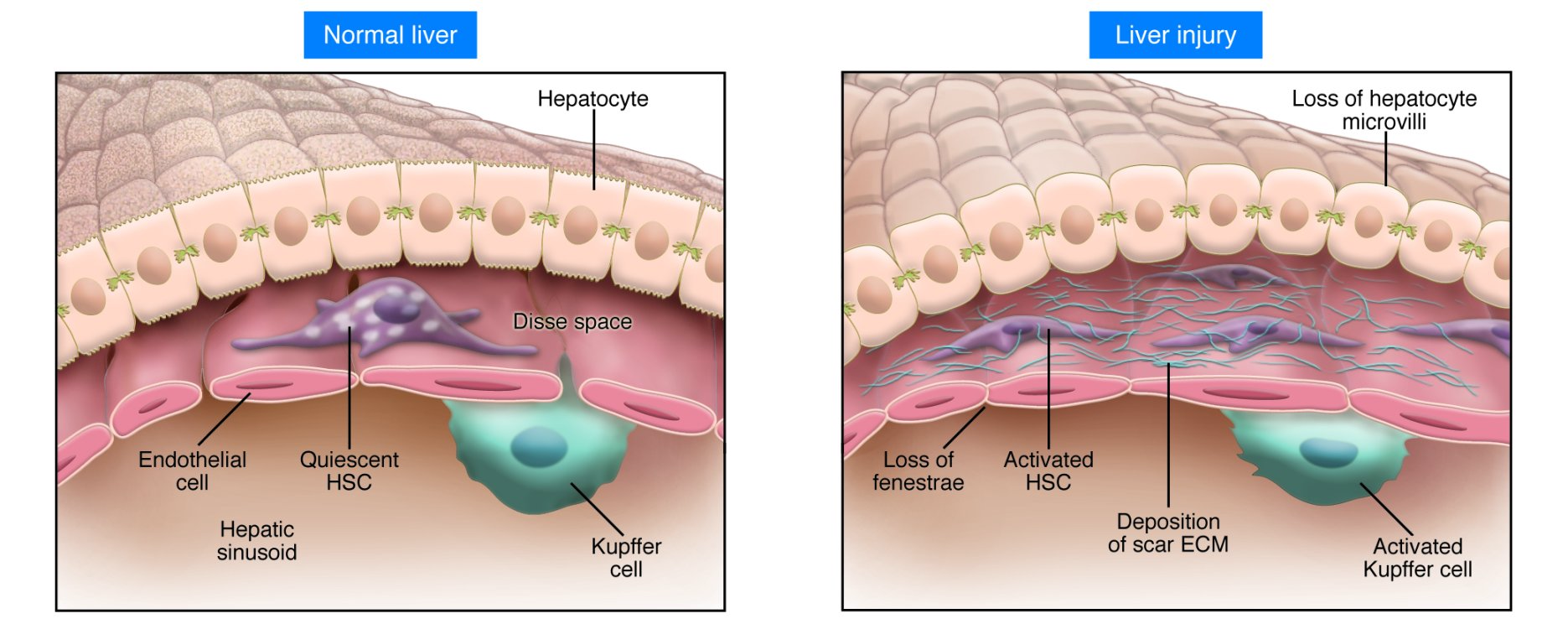 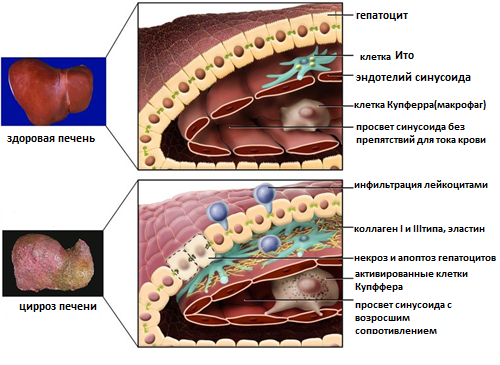 На фоне главных звеньев патогенеза цирроза ведущими клиническими проявлениями его становятся – печеночно-клеточная недостаточность и портальная гипертензия. Печёночно-клеточная недостаточностьЭто один из главных клинических синдромов при циррозе, однако, может иметь место при развитии гепатитов и гепатозов. Связана она с уменьшением массы паренхимы печени (мало «работников», а оставшиеся работаю плохо). При печеночно-клеточной недостаточности печени могут нарушаться любые или даже все функции печени. Количество нарушенных функций и их степень будет зависеть от степени активности, т.е. от масштаба некрозов и выраженности воспаления при гепатите или циррозе. Чем выше активность, тем хуже работает печень, как орган, выполняя свои многочисленные функции и наоборот. Выпадение функций и определяет почти целиком клинику гепатитов, циррозов и других заболеваний печени. I. Нарушение секреции желчи приводит к развитию: Условий для брожения и нарушения перистальтики, следствием это становятся: синдром мальабсорбции, мальдигестии – «ветер с дождем» (метеоризм и диарея). Из-за уменьшения секреции желчи кал становится менее насыщенного цвета. Нарушений всасывания витаминов D (остедистрофия), Е (нарушение фертильной функции, дегенерация сетчатки, полинейропатия), К (кровоточивость из-за уменьшения синтеза факторов свертывания), А (сухость и шелушение кожи, нарушение сумеречного зрения). II. Нарушение дезинтоксикационной «барьерной» функции, в отношении веществ образующихся в организме и поступающих из кишечника, в первую очередь аммиак. В организме существует два источника аммиака: а) все периферические ткани, где происходят процессы дезаминирования аминокислот; б) кишечник, где образование аммиака обусловлено процессами гниения микрофлорой. Кроме того, нарушается дезинтоксикация таких веществ как фенол, индол, скатол, амины (путресцин, кадаверин), соединений серы, образующиеся при окислении метионина (диметилсульфид, метилмеркаптан). Диметилсульфид и метилмеркаптан при тяжелой печеночной недостаточности обуславливают характерный печеночный запах. Нарушение дезинтоксикационной функции может быть связано с уменьшением массы функционирующих гепатоцитов, а в случае цирроза еще и шунтированием крови в обход печени по порто-кавальным анастамозам из-за портальной гипертензии Нарушения дезинтоксикационной функции печени проявляются признаками интоксикации, затрагивающими, прежде всего центральную нервную систему (печеночная энцефалопатия, кома), т.к. аммиак и другие токсичные продукты грубо внедряются и разрывают цикл три(ди-)карбоновых кислот, делая затрудненным синтез АТФ. В печени обезвреживаются некоторые эндогенные второстепенные пирогены, например, этиохоланолон, а накопление этих пирогенов может проявиться субфебрильной лихорадкой. Нарушение функции дезинтоксикации необходимо учитывать при использовании фармакологических препаратов, т.к., ухудшается метаболизм лекарственных препаратов, которые метаболизируются в печени, следовательно, такие препараты не должны использоваться у больных с заболеваниями печени или их дозы должны быть уменьшены, например фенобарбитал, парацетамол и др. III. Нарушение обмена гормонов: некоторые гормоны и БАВ подвергаются деградации в печени, отсутствие процесса их разрушения приводит к их вторичному повышению. Например, из-за развития гипергастринемии и гипергистаминемии – у пациентов с тяжелой печено-клеточной недостаточностью часто развивается язва желудка и 12-кишки. Особое внимание всегда уделяется инактивации печенью эстрогенов. Гиперэстрогенемия – приводит к появлению важных диагностических признаков, которые объединили понятием «синдром малых признаков цирроза»: карминово-красная окраска губ, малиновый язык, пальмарная эритема («руки любителя пивасика»۩), сосудистые звездочки/сосудистые паучки (появляются выше линии сосков), гинекомастия ϾϿ и галакторея, атрофия полового члена, яичек , выпадение волос, у женщин нарушение менструального цикла, либидо и фертильной функции. Вторичный гиперальдостеронизм тоже имееть место при печночно-клеточно недостаточности, он проявится: склонностью к отекам, сухостью в полости рта и жаждой, может повышаться АД. IV. Нарушение белкового обмена: гипоальбуминемия приведет к отекам из-за снижения онкотического давления, уменьшается синтез факторов свертывания (геморрагический синдром), нарушение метаболизма аммиака приводит к тяжелым нарушениям в работе цикла ди(три-)карбоновых кислот. V. Нарушения углеводного обмена. Нарушение гликогенолиза приводит к гипогликемии, которая в свою очередь приводит к активации континсулярных гормонов, в результате со временем произойдет нарушение толерантности к глюкозе или сахарный диабет. VI. Расстройства пигментного обмена: проявляются развитие паренхиматозной желтухи, которая описана выше. VII. Расстройства обмена витаминов и микроэлементов: из-за нарушения секреции желчи происходит нарушение эмульгирования жиров и всасывания жирорастворимых – D,E,K,A. Печень является местом депонирования витаминов В12, железа, меди, селена и других. Печень играет ключевую роль в метаболизме витамина В6. Все это приводит к развитию гиповитаминозов и дефициту микроэлементов. VIII. Нарушение фагоцитарной функциивыполняют особые звездчатые эндотелиальные клетки (клетки Купффера), относящиеся к системе мононуклеарных фагоцитов. Повреждение гепатоцитов (не только при циррозах, но и гепатитах, гепатозах) сопровождается увеличением клеточной проницаемости и выходом клеточных ферментов в кровь, что определяется, как цитолитический синдром. Синдром цитолиза (цитолитический синдром) - неспецифическая реакция клеток печени па действие повреждающих факторов (разрушение клеток печени). В основе его лежит изменение проницаемости мембран клеток и их органелл. Приводящее к выходу составных частей клеток в межклеточное пространство и в кровь, в частности ферментов. Принято выделять гепатонеспецифичные ферменты: АсАТ, АлАТ, ЛДГ и гепатоспецифичные ферменты (зазубри!): γ-глутаматтрансфераза, орнитинкарбамоилтрансфераза. Портальная гипертензияРазвивается в результате нарушения оттока крови из органов брюшной полости по сосудам системы воротной вены, характерный признак цирроза, но встречается и при других заболеваниях. В зависимости от того, где находится препятствие оттоку крови, выделяют следующие формы портальной гипертензии: подпеченочную — препятствие в стволе или крупных ветвях воротной вены (эмболы, сдавление опухолью); внутрипеченочную: перисинусоидальный блок – из-за избыточного фиброза и воспалительной инфильтрации в области портального тракта (тетрады); постсинусоидальный блок кровотока в печени (сдавление разветвлений воротной вены узлами регенерирующих гепатоцитов или разрастаниями фиброзной ткани); наличие артериовенозных анастомозов во внутридольковых соединительнотканных септах (сброс крови из печеночной артерии в ветви воротной вены). надпеченочную — препятствие локализовано во внеорганных отделах печеночных вен или в нижней полой вене проксимальнее места впадения в нее печеночных вен. Сюда же относят портальную гипертензию, возникающую при увеличении давления в системе нижней полой вены в условиях недостаточности правого желудочка. Основные проявления синдрома портальной гипертензии: а) варикозное расширение вен пищевода и кардиальной части желудка, геморроидальных вен; б) желудочно-кишечные кровотечения, обусловленные повреждением варикозно расширенных вен; в) расширение подкожных вен передней грудной и брюшной стенки («голова медузы»); г) сброс крови из воротной вены в полые вены в обход печени, который вызывает явления интоксикации, а в тяжелых случаях развитие экзогенной (портокавальной, или шунтовой) печеночной комы. Портальная гипертензия нередко сопровождается развитием гепатолиенального, гепаторенального и гепатопульмонального синдромов. Гепато-лиенальный синдром. Его важными составными являются спленомегалия и гиперспленизм. Спленомегалия - увеличение размеров селезенки, возникает в результате застоя крови, гиперспленизм - увеличение функциональной активности селезенки - проявляется усиленным разрушением форменных элементов крови макрофагами и увеличением их депонирования в селезенке. Гиперспленизм характеризуется анемией, лейкопенией и тромбоцитопенией. Гепаторенальный синдром. Принципиально гепаторенальный и гепатопульмональный синдромы не отличаются по патогенезу. Главная причина – нарушение метаболизма (деградации) печенью простагландинов, цитокинов. Большой вклад оказывает гиперпродукция оксида азота. Однако до конца механизмы этих синдромов не раскрыты. Проявляется нарушениями фильтрационной способности почечных клубочков при сохранности функций канальцевого эпителия. Дополнительной причиной этого синдрома является гипоальбуминемия. Гепатопульмональный синдром – легочная гипертензия частый спутник портальной гипертензии. Характерным является дилатация сосудов на фоне спазма прекапиллярных сосудов (артериол), что много кратно увеличивает гипертензиию в малом круге. Важным клиническим проявлением портальной гипертензии является асцит. АсцитАсцит – это скопление свободной жидкости (как правило, транссудата) в брюшной полости. Причинами асцита могут быть: а) портальная гипертензия разного происхождения; б) отеки при хронической недостаточности сердца, заболеваниях почек, алиментарной дистрофии; в) нарушения оттока лимфы по грудному протоку (его ранение, сдавление); г) поражение брюшины опухолевым или туберкулезным процессом (асцит-перитонит). Асцитическая жидкость по своему характеру бывает обычно серозной, значительно реже — геморрагической. В патогенезе асцита имеют значение следующие механизмы: Гидростатический - связанс повышением давления крови в капиллярах сосудов воротной системы; Онкотический - обусловлен уменьшением белоксинтетической функции печени (уменьшение синтеза альбуминов); Повышение ОЦК и задержка натрия в организме - связана с увеличением содержания альдостерона в крови. Это в свою очередь обусловлено активацией ренин- ангиотензинной системы (застой крови в сосудах воротной системы →уменьшение венозного возврата → падение минутного объема сердца→гипоксия почек →выделение ренина). Кроме того, в связи с нарушением функций печени может нарушаться инактивация альдостерона, что равнозначно его гиперпродукции; Лимфогенный механизм - в связи с нарушением лимфооттока происходит переход богатой белками лимфы в брюшную полость. Это вызывает повышение онкотического давления жидкости брюшной полости с последующим выходом в нее воды из кровеносных сосудов и интерстициального пространства. Печеночная комаЭтиология. Любые острые/хронические поражения печени. Патогенез печеночной комы Существует 2 главных варианта патогенеза печеночной комы: эндогенная – связана с тяжелой гепатоцеллюлярной недостаточностью, т.е. прямым поражением паренхимы печени; экзогенная – связана с «выключением» печеночного кровотока по v.portae в результате активации шунтов и сброса крови по анастамозам в обход печени. Как и любая кома при печеночной кома – проявление охранительного торможения коры в ответ на тяжелые метаболические расстройства, такое торможение значительно уменьшает потребность коры в АТФ (режим гибернации в компьютере). Коме предшествует печеночная энцефалопатия, печеночная прекома, патогенез которых тот же, но степень метаболических расстройств была достаточна для поддержания активности коры, хоть и на низком уровне. Патогенетические факторы печеночной комы 1. Выпадение обезвреживающей функции печени и действие токсических продуктов на печень. Основными токсинами является аммиак и метилмеркаптаны. Аммиак обезвреживается в печени, превращается в мочевину и выводится в основном с мочой. В результате нарушения метаболизма аммиака (при эндогенной коме) и/или сброса его по анастамозам в системный кровоток концентрация его в крови резко увеличивается. Это вызывает: энергодефицит нейронов из-за того что аммиак грубо вмешивается и нарушает цикл Кребса – уменьшается синтез АТФ; уменьшается синтез возбуждающих нейромедиаторов; прямое токсическое воздействие на нейроны, ингибирование ионных каналов нейронов. Кроме аммиака в крови накапливаются другие церебротоксичные вещества: метилмеркаптаны (они еще придает выдыхаемому воздуху пациента «печеночный сладкий запах»), серосодержащие аминокислоты (метионин, цистеин), продукты метаболизма триптофана (скатол, индол). 2. Усиление синтеза тормозных нейромедиаторов и образование ложных нейромедиаторов. В головном мозге больных с печеночной недостаточностью повышен уровень ГАМК – одного из главных тормозных медиаторов, а вот количество возбуждающих нейромедиаторов повышено. Известно, что главными возбуждающими нейромедиаторами являются катехоламины и дофамин. Для их синтеза необходимы аминокислоты с разветвленной цепью (валин, лейцин, изолейцин). При печеночной недостаточности наблюдается усиленный катаболизм белка и использование в качестве энергетического источника аминокислот с разветвленной цепью, что приводит к одновременному накоплению ароматических аминокислот. Дополнительно ароматические аминокислолты накапливаются из-за расстройств дезаминирования. В результате соотношение аминокислот: 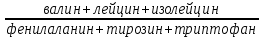 которое необходимо для синтеза возбуждающих медиаторов, смещается в сторону уменьшения, в сторону увеличения знаменателя – ароматических аминокислот. Последние являются предшественниками ложных нейромедиаторов – октопамина, тирамина. Ложные нейромедиаторы конкурируют с нормальными возбуждающими медиаторами головного мозга – норадреналином, дофамином, адреналином – в результате ложные медиаторы связываются с сайтами возбуждающих рецепторов, инактивируют их, что отражается угнетением нервной системы. 3. Нарушения КОС. В начале развивается метаболический ацидоз из-за накопления в крови ПВК и лактата, которые вызывают активацию дыхательного центра, развитие гипервентиляции, которая переводит метаболический ацидоз в респираторный алкалоз. Гипокалиемия может привести к развитию и метаболического алкалоза. Расстройства КОС вызывают повышение проницаемости гематоэнцефалического барьера, что усиливает в свою очередь проникновение аммиака к нейронам. 4. Электролитные нарушения. При печеночной недостаточности нарушается метаболизм гормонов в печени, в том числе альдостерона, развивается вторичный гиперальдостеронизм и, как следствие, гипокалиемия и гипернатриемия. Гипокалиемия усугубляет алкалоз, способствует переходу натрия и ионов водорода в клетку, следствием чего является внутриклеточный отёк и ацидоз, который усиливается энергодефицитом на фоне повышения концентрации аммиака. 5. Гипоксия смешанного генеза: тканевая из-за внедрения аммиака и других токсичных продуктов в цикл Кребса, циркуляторная из-за падения тонуса сосудодвигательного центра и ДВС синдрома. 6. ДВС синдром. Токсические продукты, ацидоз, электролитные расстройства приводят к высвобождению тканевого тромбопластина из гепатоцитов и других клеток в большом объеме, что запускает повсеместную активация коагуляционного гемостаза и запуск ДВС синдрома. Имеет значение и дефицит антитромбина III из-за снижения его синтеза печенью на фоне недостаточности функций печени. 7. Поражение почек развивается в результате гепаторенального синдрома, а также поражения коркового вещества почек на фоне эндотоксемии, гипоксии, ДВС-синдрома. Это приводит к развитию почечной недостаточности и усугублению метаболических расстройств. В прекоматозном состоянии в период печеночной энцефалопатии у пациентов появляется изменения в личности: эгоцентризм, уменьшение интереса к семье, нестабильное настроение, ребячество. С неврологической стороны наблюдается инверсия сна (дисхроноз), апатия, сонливость, изменения почерка, невнятная речь, астериксис (без обеликсис ). В дальнейшем неврологические изменения усугубляются, сильнее нарушается сознание вплоть до ступора и сопора, при отсутствии вмешательства пациент впадает в кому с тяжелым угнетением ЦНС. Итогом выше перечисленных патогенетических путей становится полиорганная, которая приводит к смерти. ГЕПАТИТЫХронический неактивный гепатит — воспаление печени, не выходящее за пределы портальной триады, без фиброза. Биопсия печени выявляет портальную лимфоцитарную инфильтрацию. Клинически у большинства больных течение бессимптомное, доброкачественное. Возможны утомляемость, анорексия и боль в животе. Хронический активный гепатит — воспаление печени, выходящее за пределы портальной зоны, с признаками ступенчатого некроза и фиброза. Заболевание часто прогрессирует в цирроз печени. Клинически: гепатомегалия, сосудистые «звёздочки», синдром гиперспленизма и гаммапатии, гиповитаминоз, нарушения функции эндокринной системы, внепечёночные проявления и другие признаки хронического заболевания печени. Фиброз— важная характеристика хронического гепатита; его выраженность различна: от минимальной до развитого цирроза. При незначительном фиброзе происходит развитие соединительной ткани вокруг сосудов портальных зон. При выраженных формах фиброзная ткань захватывает периферию долек, гнёзда гепатоцитов при этом оказываются в окружении соединительной ткани. При прогрессировании процесса соединительнотканные тяжи простираются до центральной вены. Значительная гибель гепатоцитов и их замещение соединительной тканью с нарушением гистоархитектоники и микроциркуляции, по сути дела, означают цирроз печени — частый исход хронического гепатита. Осложнения: хроническая печёночная недостаточность и портальная гипертензия. |