реферат по биологии. Реферат. Галактоземия
 Скачать 228.36 Kb. Скачать 228.36 Kb.
|

|
МИНЗДРАВ РОССИИ Федеральное государственное бюджетное образовательное учреждение высшего образования «Южно-Уральский государственный медицинский университет» Министерства здравоохранения Российской Федерации (ФГБОУ ВО ЮУГМУ Минздрава России) Кафедра Биологии РЕФЕРАТ Тема: «Галактоземия» Выполнила: Липатова Полина Григорьевна Группа № 113, лечебный факультет «9» декабря 2021г. Проверил : заведующий кафедрой, д.м.н. Колесников Олег Леонидович «9» декабря 2021г Челябинск 2021 год ОглавлениеВведение Актуальность темы: Наследственные заболевания - болезни, обусловленные нарушениями в процессах хранения, передачи и реализации генетической информации. Доля наследственных заболеваний составляет около 10% всех заболеваний человека. С развитием генетики человека, в том числе и генетики медицинской, выяснилась наследственная природа многих заболеваний и синдромов, считавшихся ранее болезнями с неустановленной этиологией. Роль наследственных факторов подтверждается более высокой частотой ряда заболеваний в некоторых семьях по сравнению с населением в целом. Изучением наследственных заболеваний человека занимается преимущественно медицинская генетика. В их основе лежат мутации — хромосомные и генные, соответственно чему условно говорят о хромосомных болезнях и собственно наследственных (генных) болезнях. Мутация ведёт к нарушению синтеза определенного полипептида (структурного белка или фермента). В зависимости от того, какова роль этого полипептида в жизнедеятельности организма, у больного возникают нарушения (изменения фенотипа) локального или системного порядка. Одним из примеров наследственных заболеваний является галактоземия. Мне стало интересно узнать больше о данном заболевании. Как его диагностируют? Какие методы используют для лечения данного заболевания в нашей стране? На эти и другие вопросы я попыталась ответить в своей работе. Цель работы: изучить генетические болезни на примере галактоземии и разобрать симптоматику и способы лечения данного заболевания. Задачи: -ознакомиться с классификацией и патогенезом галактоземии; -изучить частоту встречаемости данного заболевания; -ознакомиться с клинической картиной галактоземии; -узнать о возможных методах диагностики, лечения и профилактики галактоземии. Основная часть Определение: Галактоземия – наследственное нарушение обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов (галактозо-1-фосфата и галактитола), что обусловливает клиническую картину заболевания и формирование отсроченных осложнений. Тип наследования галактоземии - аутосомно-рецессивный. КОД МКБ 10: E74.2 - Галактоземия (нарушения обмена галактозы; включена недостаточность галактокиназы). Эпидемиология: Частота галактоземии по данным массового обследования новорожденных в России составляет 1: 20 000, при этом подавляющее большинство случаев заболевания обусловлено мутациями в гене GALT. Этиология: Галактоза (от греческого слова gala, galaktos - молоко) представляет собой моносахарид - C-4 эпимер глюкозы, с идентичной молекулярной формулой, но с отличной от глюкозы структурной формулой. Несмотря на большое сходство молекул глюкозы и галактозы, превращение последней в глюкозу требует нескольких эволюционно-консервативных ферментативных реакций, которые протекают в цитоплазме клетки и известны под названием пути Лелуара метаболизма галактозы (рисунок 1). Галактоза имеет важнейшее значение для роста и развития детского организма, так как является компонентом пищи грудного ребенка, входя в состав молока. 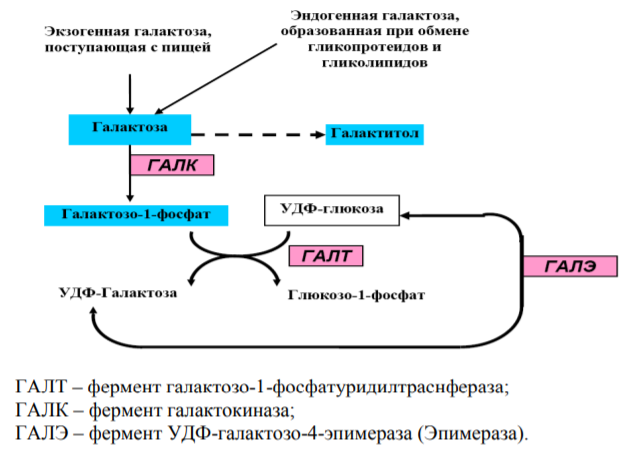 Рисунок 1 – Общая схема обмена галактозы. Галактоземия относится к наследственным болезням углеводного обмена и объединяет несколько генетически гетерогенных форм. В основе заболевания лежит недостаточность одного из трех ферментов, участвующих в метаболизме галактозы: галактозо-1-фосфатуридилтраснферазы (ГАЛТ), галактокиназы (ГАЛК) и уридин-дифосфат(УДФ)-галактозо-4-эпимиразы (ГАЛЭ) (рисунок 1). Известны три гена, мутации в которых могут приводить к развитию галактоземии (таблица 1). Таблица 1 – Генетическая гетерогенность галактоземии.
Патогенез: Галактоза играет крайне важную роль в организме, особенно растущего ребенка. Этот моносахарид не только является значимым источником энергии для клетки, но и служит необходимым пластическим материалом для образования гликопротеидов, гликолипидов и других комплексных соединений, используемых организмом для формирования клеточных мембран, нервной ткани, нервных окончаний, процессов миелинизации нейронов и др. Нарушение метаболизма галактозы, наблюдаемое при галактоземии, неизбежно приводит к расстройству функционирования многих органов и систем организма. Основным источником галактозы у человека является пища. Большое количество потребляемых в течение дня пищевых продуктов содержат лактозу, из которой в кишечнике в результате гидролиза образуется галактоза; многие продукты питания содержат галактозу в чистом виде. У человека этот моносахарид также может образовываться и эндогенным путем, подавляющее его количество получается в процессе ферментативных реакций между уридиндифосфатглюкозой (УДФ-глюкозой) и УДФ-галактозой, а также в процессе обмена гликопротеинов и гликолипидов. Патогенетические механизмы галактоземии до сих пор полностью не изучены. В результате недостаточности любого из трех ферментов – ГАЛТ, ГАЛК или ГАЛЭ – в крови повышается концентрация галактозы. При дефиците активности ферментов ГАЛТ и ГАЛЭ, помимо избытка галактозы, в организме больного накапливается также избыточное количество галактозо-1-фосфата, что на сегодняшний день считается основным патогенетическим фактором, обусловливающим большинство клинических проявлений галактоземии и формирование отсроченных осложнений. Избыток галактозы в организме может метаболизироваться другими биохимическими путями: в присутствии НАДФ·Н (или НАД·Н) она может превращаться в галактитол. Накопление галактитола в крови и тканях и повышение его экскреции с мочой наблюдается при всех формах галактоземии; в хрусталике глаза избыток галактитола способствует формированию катаракты. Имеются сведения о том, что высокое содержание галактитола в тканях мозга способствует набуханию нервных клеток и формированию псевдоопухоли мозга у отдельных больных. Патологические процессы при галактоземии обусловлены не только токсическим действием указанных продуктов, но и их тормозящим влиянием на активность других ферментов, участвующих в углеводном обмене (фосфоглюкомутазы, глюкозо-6-фосфатдегидрогеназы), следствием чего является гипогликемический синдром. Предполагается также, что предрасположенность к сепсису у новорожденных с галактоземией обусловлена ингибированием бактериальной активности лейкоцитов. Классификация: В основу современной классификации галактоземии положен этиологический принцип. Существуют три типа галактоземии, в зависимости от имеющегося у больного дефекта одного из трех основных ферментов, участвующих в метаболизме галактозы: Классическая - галактоземия I типа, обусловленная дефицитом фермента галактозо-1-фосфат-уридилтрансферазы (ГАЛТ). Этот тип галактоземии также включает в себя вариант Дуарте. Недостаточность галактокиназы (ГАЛК) (галактоземия II типа). Дефицит уридиндифосфат-галактозо-4-эпимеразы (ГАЛЭ или эпимеразы) – галактоземия III типа. Клиническая картина: Классическая галактоземия - наиболее тяжелая форма нарушения метаболизма галактозы, вызывается дефицитом активности фермента ГАЛТ. Заболевание обычно манифестирует в первые дни - недели жизни, быстро прогрессирует и в отсутствии лечения носит жизнеугрожающий характер. На фоне вскармливания молоком у новорожденного появляется рвота, диарея, мышечная гипотония, сонливость, вялость. Останавливается прибавка в массе тела, наблюдается вялое сосание, появляются и нарастают признаки поражения печени, часто сопровождающиеся гипогликемией, желтухой и гепатомегалией; нередко отмечается кровоточивость в связи с гипокоагуляцией; у многих больных возникает нарушение функции канальцев почек. Наиболее тяжелым проявлением галактоземии у новорожденных является сепсис, который имеет фатальное течение и чаще всего обусловлен грамположительными микроорганизмами, в 90% случаев - Escherichia coli. Если не оказывать своевременную медицинскую помощь, то около 75% больных в младенческом возрасте умирают. Приблизительно 20-30% детей с классической формой этого заболевания погибают от сепсиса, также высок процент смертности детей первых месяцев жизни от хронической недостаточности функции печени. Уже в первые несколько дней после рождения у ребенка нередко можно диагностировать катаракту. Иногда катаракта выявляется не в результате офтальмоскопии, а при исследовании на щелевой лампе, так как она состоит из точечных помутнений в фетальном ядре хрусталика. Лабораторные исследования выявляют повышение активности трансаминаз в сыворотке крови и повышение концентрации билирубина. Неконъюгированная гипербилирубинемия, характерная для ранней стадии болезни, далее переходит в конъюгированную. В биохимических анализах крови часто определяется гипогликемия и гипоальбуминемия, может быть повышенным содержание аммиака и аминокислот, особенно фенилаланина, тирозина и метионина. Наблюдается снижение синтеза факторов свертывания крови. Нередко обнаруживается гиперхлоремический метаболический ацидоз, гипофосфатемия, генерализованная гипераминацидурия, что свидетельствует о вторичной дисфункции канальцев почек. Ультразвуковое исследование выявляет увеличение размеров печени. Наличие воспалительных изменений в анализах периферической крови в сочетании с повышением уровня Среактивного белка и положительным прокальцитониновым тестом является критерием присоединения сепсиса. У выживших детей, не получавших адекватного лечения, развивается хроническая печеночная недостаточность и тяжелое поражение нервной системы с резким отставанием психомоторного развития, что приводит к глубокой инвалидизации и уменьшению продолжительности жизни. Степень выраженности клинических проявлений галактоземии зависит от величины снижения активности фермента ГАЛТ. При классической галактоземии активность галактозо-1-фосфатуридилтрансферазы не превышает 5% от нормальных значений. Существует несколько вариантов частичного дефицита ГАЛТ. Наиболее частый из них – вариант Дуарте, при котором у больных имеется один аллель Дуарте (несущий определенную мутацию в гене GALT, приводящую к незначительному снижению активности фермента, этот аллель принято обозначать символом D) и аллель классической галактоземии (обозначается символом G). У больных, имеющих генотип D/G, активность ГАЛТ составляет 5 – 25% от нормы; у больных, имеющих два аллеля Дуарте (D/D), активность фермента равна приблизительно 25%. У таких детей в периоде новорожденности обычно не наблюдается тяжелых жизнеугрожающих клинических проявлений. Однако часто обращает на себя внимание длительная желтуха (в течение первых 2 месяцев), увеличение размеров печени, ультразвуковые признаки фиброза печени. Наряду с этими симптомами может наблюдаться плохая прибавка в массе тела, задержка физического развития, темповая задержка моторного развития. С первых месяцев жизни у многих детей обнаруживается катаракта. У тех лиц, у которых активность ГАЛТ выше 50% от нормы, зачастую какие-либо клинические признаки галактоземии 12 отсутствуют или носят несущественный характер и не требуют специального лечения. Галактоземия II типа, связанная с недостаточностью фермента галактокиназы (ГАЛК). Этот тип галактоземии встречается значительно реже, частота его неизвестна, но вероятно, составляет менее 1:100 000. Клиническая симптоматика при галактоземии II типа менее яркая, чем при классическом варианте заболевания. Единственным проявлением дефицита ГАЛК у многих больных может быть формирование катаракты. Такие симптомы заболевания как отставание роста и массы тела, диспептические расстройства и др., незначительно выражены, хотя и могут наблюдаться у детей грудного возраста, когда они начинают употреблять в пищу значительное количество молока. Более информативными симптомами являются: галактозурия, гипергалактоземия и постепенно развивающаяся катаракта. Описаны лишь единичные случаи задержки психомоторного развития у детей с галактоземией, связанной с недостаточностью галактокиназы. Галактоземия III типа (недостаточность фермента уридиндифосфатгалактозо-4-эпимеразы - ГАЛЭ). Этот тип галактоземии встречается исключительно редко, частота его точно неизвестна. Выделяют 2 клинические формы галактоземии, связанные с недостаточностью ГАЛЭ: доброкачественную (изолированную) и тяжелую (генерализованную). Доброкачественная форма (или периферическая) характеризуется дефицитом энзима только в циркулирующих клетках крови, в то время как при тяжелой форме дефицит энзима определяется во всех тканях. Клинические проявления при доброкачественной форме могут отсутствовать, и заболевание выявляется случайно при обнаружении повышенного уровня галактозы в крови (при проведении скрининга новорожденных, биохимическом анализе крови и др.). При этом уровень активности фермента понижен только в клетках периферической крови, в то время как в печени, культуре фибробластов и ктивированных лимфоцитах сохраняется на нормальных значениях. Однако катамнестические наблюдения показывают, что у части детей в последующем имеются нарушения моторных функций и речевого развития. При тяжелой форме заболевания начальные симптомы имеют сходство с классической галактоземией, включая желтуху, рвоту, мышечную гипотонию, задержку развития, гепатомегалию, умеренную гипераминоацидурию, и значительную галактозурию. В последующем наблюдается увеличение селезенки. Несмотря на раннее распознавание заболевания и включение диетического лечения, через 2-3 года нередко обнаруживаются отчетливая задержка психомоторного развития и нейросенсорная глухота. Активность фермента снижена не только в эритроцитах, но также в клетках печени и фибробластах кожи. Некоторые больные составляют группу риска по развитию катаракты из-за нарушенного обмена галактозы. Внедрение в Российской Федерации неонатального скрининга на галактоземию позволяет выявить это заболевание в периоде новорожденности практически у 100% больных. Однако в тех случаях, когда проведение скринингового исследования по каким-либо причинам оказалось невозможным или были получены ложноотрицательные результаты, следует исключать галактоземию у тех детей, у которых имеются не только типичные вышеописанные проявления классической галактоземии, но и такие признаки как: - сочетание желтухи и геморрагического диатеза в первые 2 недели жизни; - появление катаракты в раннем возрасте, особенно на первом году жизни; - сепсис у доношенного новорожденного, вызванный E. coli; - сочетание задержки прибавки в массе тела, увеличения размеров печени, мышечной гипотонии, задержки моторного развития и катаракты у детей раннего возраста. Диагностика: В соответствии с приказом Минздравсоцразвития РФ № 185 от 22.03.2006 года «О массовом обследовании новорожденных детей на наследственные заболевания», в Российской Федерации массовый неонатальный скрининг на галактоземию проводят на 4-е сутки жизни доношенным новорожденным и на 7-е сутки жизни - недоношенным детям. Забор образца крови производят из пятки новорожденного на фильтровальный бланк. Внедрение программы неонатального скрининга позволяет выявить галактоземию практически у 100% детей с этим заболеванием. При помощи флуоресцентного метода проводят определение уровня тотальной галактозы в пятнах высушенной крови, который представляет собой сумму концентрации галактозы и галактозо-1-фосфата. При выявлении уровня тотальной галактозы 7,2 мг/дл и выше проводится подтверждающая диагностика классической галактоземии, которая включает в себя определение активности фермента ГАЛТ и ДНК-диагностику с целью выявления наиболее распространенных мутаций в гене GALT и полиморфного варианта Дуарте, приводящего к снижению активности фермента. Определение активности ГАЛТ проводят в пятнах высушенной крови и/или в цельной гепаринизированной крови, ДНК-исследование осуществляют в образце цельной крови. Взятие образца крови для ДНК-исследования производят из периферической вены с добавлением в пробирку консерванта – ЭДТА или гепарина. Диагностически значимым считается снижение активности фермента ГАЛТ в высушенных пятнах крови < 2,5 Е/гHb, в цельной крови - < 1,3 Е/гHb. Подтверждающая ДНК-диагностика классической галактоземии включает в себя два этапа: 1) скрининг на наиболее частые мутации в гене GALT и вариант Дуарте; 2) полный анализ гена методом прямого автоматического секвенирования для выявления более редких мутаций. На первом этапе наиболее целесообразно проведение поиска следующих мутаций в гене GALT: р.Gln188Arg, p.Lys285Asn, IVS3-2a->c, p.Met142Lys, p.Leu358Pro, составляющие в совокупности 82,1% мутантных аллелей в российской популяции, и p.Asn314Asp (N314D, вариант Дуарте). Алгоритм диагностики классической галактоземии представлен на рисунке 2. 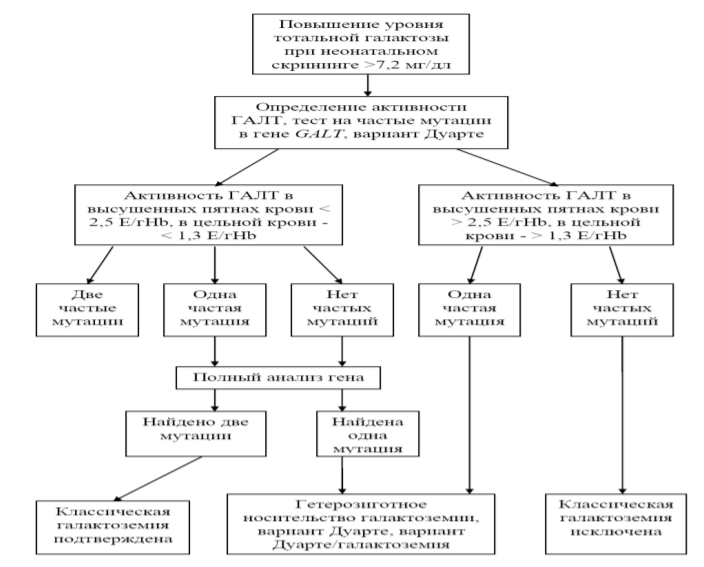 Рисунок 2 – Алгоритм подтверждающей диагностики галактоземии I типа. При наличии у ребенка клинических проявлений классической галактоземии в сочетании с положительным результатом неонатального скрининга на галактоземию и нормальной активностью фермента ГАЛТ проводится поиск мутаций в гене GALE, кодирующем фермент уридиндифосфат-галактозо-4-эпимеразу (ГАЛЭ), для исключения галактоземии III типа. Дифференциальная диагностика галактоземии проводится с: - болезнями, сопровождающимися повышенным выделением сахара (меллитуриями, сахарным диабетом, цистинозом, синдром Фанкони); - болезнями, сопровождающимися увеличением печени и желтухой (гепатит, токсоплазмоз, гликогенозы, синдром Криглера-Найара, недостаточность α– антитрипсина, тирозинемия, тип Ι, синдром Фанкони-Бикеля, болезнь НиманаПика тип С, болезнь Вильсона, дефицит печеночного белка - цитрина, печеночная гемангиоэндотелиома и др.); - врожденными аномалиями желчевыводящей системы (открытый аранциев проток, портокавальный шунт -между воротной веной и нижней полой веной, гипоплазия внутрипеченочной портальной вены, обструкции билиарного тракта, в том числе синдром Алажиля; прогрессирующий внутрипеченочный холестаз (болезнь Byler); - транзиторной гипергалактоземией, которая отмечается при позднем закрытии венозного протока (Ductus venosus) и исчезает в течение 2-3-5 месяцев после его закрытия, при гетерозиготностном носительстве мажорных мутаций в генах GALT и GALE. Поэтому для проведения дифференциальной диагностики требуется проведение углубленного инструментально-лабораторного обследования: допплеровского УЗИ-исследования печени и желчевыводящих путей, исследование альфа-фетопротеина и уровня желчных кислот в крови, которые могут быть повышены при порто-кавальном анастомозе и дефиците цитрина) и др. Лечение: При получении положительных результатов неонатального скрининга назначается диетотерапия, одновременно определяется активность фермента ГАЛТ и проводится молекулярно-генетическое обследование. Основным методом лечения при галактоземии является диетотерапия, предусматривающая пожизненное исключение из рациона продуктов, содержащих галактозу и лактозу. Необходимо полностью исключить из рациона больного любой вид молока (в том числе женское, коровье, козье, детские молочные смеси и др.) и все молочные продукты, а также строго избегать употребления тех продуктов, куда они могут добавляться (хлеб, выпечка, сосиски, колбасы, карамель, сладости, маргарины и т.п.). Запрещается также использование низколактозных смесей и молока. Ряд продуктов растительного происхождения содержит олигосахариды - галактозиды (раффинозу, стахиозу), животного происхождения - нуклеопротеины, которые могут быть потенциальными источниками галактозы (таблица 2). Таблица 2 – Продукты, содержащие галактозиды, и богатые нуклеопротеинами
В настоящее время диета с максимально строгим исключением галактозы/лактозы - это единственный способ сократить накопление токсичного компонента - галактозо-1-фосфата в тканях больного с классической галактоземией и галактитола у больных с дефицитом галактокиназы. При галактоземии, обусловленной дефицитом УДФ-Э, возможно использование низкогалактозной диеты в соответствии с допустимыми количествами галактозы в рационе под контролем ее уровня в сыворотке крови. При составлении лечебных рационов для детей первого года жизни количество основных пищевых ингредиентов и энергии должно быть приближено к физиологическим нормам. Для лечения больных галактоземией используются специализированные смеси на основе изолята соевого белка, гидролизатов казеина, безлактозные казеинпредоминантные молочные смеси, а также смеси на основе синтетических аминокислот. Лечебными продуктами первоочередного выбора для больных первого года жизни с галактоземией являются смеси на основе изолята соевого белка, в которых полностью отсутствуют растительные галактозиды. При использовании соевых смесей в питании грудных детей возможно появление аллергических реакций на белок сои. В таких случаях целесообразно назначать смеси на основе гидролизатов казеина. В зависимости от состояния ребенка возможно сочетанное применение соевой смеси и смеси на основе гидролизата казеина в соотношении 1 : 1. Возможно применение казеинпредоминантных безлактозных молочных смесей. Безлактозные молочные смеси, содержащие в составе белкового компонента 60% сывороточных белков, не должны использоваться для диетотерапии у детей с галактоземией грудного возраста, так как могут содержать следовые количества галактозы. Специализированные безлактозные/безгалактозные смеси вводят в питание больного с галактоземией в течение одного дня, сразу после установления диагноза. Прикорм вводится в период от 4 до 6 месяцев жизни. В питании используются только безмолочные продукты и блюда: безмолочные каши, которые разводят безлактозными/безгалактозными смесями, а также овощное, мясное и фруктовое пюре. Все молочные продукты, включая цельное коровье молоко, кисломолочные продукты, творог, запрещаются к использованию у больных галактоземией. Вид первого прикорма определяется состоянием желудочно-кишечного тракта и нутритивным статусом ребенка. Это может быть овощное пюре из натуральных овощей или плодоовощных консервов для детского питания без добавления молока (и не имеющих в составе бобовых) или безмолочные каши на основе кукурузной, рисовой или гречневой муки, для разведения которых необходимо использовать ту специализированную смесь, которую получает ребенок. Мясной прикорм вводят в питание с 6 месяцев. Преимущество отдают специализированным детским мясным консервам промышленного выпуска, не содержащим молока и его производных (кролик, цыпленок, индейка и др.) При выборе продуктов прикорма промышленного производства ориентируются на содержание в них галактозы (при наличии маркировки на этикетке): безопасными считаются продукты с содержанием в них галактозы не более 5 мг на 100 г продукта. При наличии галактозы в количестве от 5 до 20 мг на 100 г продукт применяется с осторожностью, под контролем содержания общей галактозы в сыворотке крови; при содержании галактозы более 20 мг в 100 г продукт не используется. При проведении симптоматической терапии следует обращать внимание на содержание лактозы в лекарственных средствах, поскольку она нередко используется в качестве вспомогательного вещества. Противопоказано применение всех гомеопатических препаратов (при их производстве используется лактоза), а также настоек и спиртовых лекарственных форм (этанол тормозит элиминацию галактозы из печени). При развитии клинической картины классической галактоземии у новорожденного может потребоваться лечение желтухи, сепсиса, нарушений функции печени и почек, центральной нервной системы. Поддерживающая терапия обычно включает в себя стандартные общепринятые мероприятия: внутривенное введение жидкости для борьбы с обезвоживанием, поддержание нормального уровня глюкозы в крови, антибиотикотерапию, лечение гипокоагуляции, заменное переливание крови. Учитывая пожизненное исключение из питания больных галактоземией молочных продуктов, для профилактики развития остеопороза, рекомендуется прием в возрастных дозировках препаратов кальция и витамина D, не содержащих лактозу. Профилактика: Профилактические меры включают медико-генетическое консультирование. В семье, где есть больной ребенок, имеется 25% риск повторного рождения больного при каждой последующей беременности. У родственников из группы риска также возможно выявление мутаций, если они были идентифицированы у больного ребенка в данной семье. Пренатальная диагностика заключается в проведении кордоцентеза, биопсии хориона в 10-12 недель гестации, амниоцентеза – в 15-18 недель гестации. Дородовая диагностика галактоземии осуществляется путем определения активности галактозо-1-фосфатуридилтрансферазы (ГАЛТ) в культуре амниоцитов, биоптате и культуре хориона, а также методами ДНКанализа, позволяющего выявлять мутации в генах GALT и GALE. Вопрос о проведении пренатальной диагностики галактоземии должен быть всесторонне обсужден с заинтересованными супружескими парами прежде, чем принять окончательное решение, так как заболевание поддается лечению. Исходы и прогноз: Прогноз заболевания неблагоприятный при поздно диагностированной тяжелой форме галактоземии (в связи с отсутствием проведения скрининга). При раннем назначении диетического лечения дети могут развиваться нормально. У некоторых больных с галактоземией сохраняется риск формирования отсроченных осложнений – задержки физического развития, нарушения развития речи («вербальной диспраксии»), моторных функций (атаксия), остеопороза. У девочек повышен риск нарушения полового созревания. Задержка психического развития, выявляющаяся у некоторых детей с галактоземией, получающих диетическое лечение, не тяжелая; она может 25 обнаруживаться в раннем возрасте, но чаще становится очевидной в школьные годы, когда у ребенка возникают трудности с чтением или усвоением математики. У части детей возможно умеренное снижение интеллектуального развития или легкая умственная отсталость. У некоторых детей школьного возраста отмечаются двигательные нарушения в виде атаксии, неловкости мелкой моторики, расстройств равновесия и координации; мышечной дистонии, возможно наличие тремора при выполнении целенаправленных действий. Задержка речевого развития у больных галактоземией в раннем возрасте может проявляться затруднением артикуляции, обедненным словарным запасом. Катаракты у больных галактоземией характеризуются как малые, транзиторные или неонатальные и проходят на фоне безлактозной/безгалактозной диеты; однако, в отдельных поздно диагностированных случаях требуется применение хирургического лечения. У больных с галактоземией наблюдается снижение минерализации костной ткани при отсутствии приема препаратов кальция и витамина D, что часто приводит к развитию раннего остеопороза и повышенному риску переломов костей. У многих лиц женского пола, больных галактоземией, развиваются признаки дисфункции яичников, может наблюдаться первичная и вторичная аменорея или олигоменорея. Заключение С развитием генетики произошли колоссальные изменения в подходах, оценке, доступности и качестве жизни пациентов с наследственными заболеваниями. Выяснилась наследственная природа многих заболеваний, считавшихся ранее болезнями с неустановленной этиологией, в том числе и галактоземии. Роль наследственных факторов подтверждается более высокой частотой ряда заболеваний в некоторых семьях по сравнению с населением в целом. Следует помнить о профилактике галактоземии. Заболевание, рассмотренное в данной работе передается по аутосомно-рецессивному типу. В семье, где есть больной ребенок, имеется 25% риск повторного рождения больного при каждой последующей беременности. У родственников из группы риска также возможно выявление мутаций, если они были идентифицированы у больного ребенка в данной семье. Пренатальная диагностика с использованием кордоцентеза для быстрой диагностики способствует своевременному отграничению заболеваний и возможному нормальному функционированию. Несмотря на то, что галактоземия относится к редким заболеваниям и диагностируется в результате проведения неонатального скрининга, иногда могут встречаться случаи, когда дети по каким-либо причинам не были обследованы на галактоземию или информация о больном ребенке получена несвоевременно. Поэтому важно знать основные аспекты диагностики, клинических проявлений, наблюдения и лечения детей с этой наследственной патологией обмена углеводов. На исход и течение заболевания влияют сроки установления диагноза своевременно и адекватно назначенная диетотерапия и мероприятия неотложной помощи (переливание крови, компонентов крови, инфузионная терапия). Прогноз заболевания неблагоприятный при поздно диагностированной тяжелой форме галактоземии (в связи с отсутствием проведения скрининга). При раннем назначении диетического лечения дети могут развиваться нормально. Список литературы Баранов А.А., Намазова-Баранова Л.С., Боровик Т.Э., Ладодо К.С., Бушуева Т.В., Маслова О.И., Кузенкова Л.М., Журкова Н.В., Звонкова Н.Г. и др.. Диетотерапия при наследственных болезнях аминокислотного обмена Методическое письмо. Москва. 2013. 97 с. Волгина С.Я., Асанов А.Ю., Соколов А.А. Современные аспекты диагностики, лечения и наблюдения детей с галактоземией I типа. Российский вестник перинатологии и педиатрии. 2015;60(5):179-187. Краснопольская К.Д. Наследственные болезни обмена веществ. М., 2005; 364. Захарова Е.Ю., Воскобоева Е.Ю., Байдакова Г.В. Галактоземия, тип 1: клинические проявления, диагностика и лечение. В кн. «Молекулярно-биологические технологии в медицинской практике». Новосибирск: Альфа Виста, 2006; 155–60 Гинтер Е.К., Наследственные болезни / под ред. Е.К. Гинтера, В.П. Пузырева - М. : ГЭОТАР-Медиа, 2017. - 464 с. - ISBN 978-5-9704-3969-2 Акуленко Л. В. Медицинская генетика: учебник / Л. В. Акуленко, И. В. Угаров ; под ред. О. О. Янушевича, С. Д. Арутюнов. - Москва : ГЭОТАР-Медиа, 2011. - 208 с. Хандогина Е. К. [и др.]. Генетика человека с основами медицинской генетики: учеб. - 2-е изд., перераб. и доп. - Москва : ГЭОТАР-Медиа, 2014. - 192 с. |