ТЕМА: КАТАЛИЗ ХИМИЧЕСКИХ ПРОЦЕССОВ
СОДЕРЖАНИЕ
Введение
1. Сущность явления катализа и основные понятия каталитической химии.
2. Основные типы катализаторов и механизмы каталитических реакций.
3. Место каталитической химии в системе химических знаний.
Введение
Открытие явления катализа без сомнения следует отнести к величайшим достижениям химической науки, к важнейшему этапу в создании современной техники и эффективных технологий – цивилизации ХХ века. Явление катализа – основа существования живой клетки и, как полагают, могло иметь решающее значение в процессе возникновения жизни. Без каталитической химии сегодня трудно представить химическую промышленность, в которой более 90% всех процессов – каталитические процессы.
Биологические катализаторы – ферменты человек использовал на ранних стадиях своего развития в процессах получения вина, винного уксуса, винного спирта, при изготовлении сыров. Первые сообщения о синтезах серного эфира и этилена из этанола с применением кислотных катализаторов относятся к XVI – XVII векам[1] . Вместе с тем, историю кислотного катализа принято отсчитывать от классической работы К. Кирхгофа по сернокислотному гидролизу крахмала, результаты которой он доложил в 1811 году в Российской Академии наук.
Началом сознательного применения металлов для ускорения химических реакций считают работы Л. Тенара, братьев Дэви и И. Деберайнера (1813 – 1823). История катализа комплексами тяжелых металлов начинается с открытого М.Г. Кучеровым в 1881 году катализа реакции гидратации ацетилена солями ртути.
Первые обобщения фактов каталитического действия были сделаны Л. Митчерлихом и Й.Я. Берцелиусом в 1834 – 1835 гг. С этого момента явление катализа стало объектом науки и основой каталитических методов проведения химических реакций[2] .
1. СУЩНОСТЬ ЯВЛЕНИЯ КАТАЛИЗА И ОСНОВНЫЕ ПОНЯТИЯ КАТАЛИТИЧЕСКОЙ ХИМИИ.
Для объяснения явления катализа уже в первых теориях привлекались представления об особой “каталитической силе”, о химической природе катализа (образование промежуточных химических соединений) и о роли физических факторов (“сгущение” молекул на поверхности твердых тел).
Явление катализа настолько поражало химиков, что для его объяснения (особенно в случае катализа твердыми телами) пытались найти какие-то особые свойства твердых тел: активные центры на поверхности (Х.С. Тэйлор, 1925–1930 гг.), дублеты и мультиплеты поверхностных атомов с их геометрическим и энергетическим соответствием реагирующим молекулам (А.А. Баландин, 1929 – 1967 гг.), полупроводниковые свойства твердых тел (К. Хауффе, С.З. Рогинский, Ф.Ф. Волькенштейн, 1938 – 1950 гг.).
Несмотря на то, что такие исследователи, как В. Оствальд, П. Сабатье и В.Н. Ипатьев (конец XIX – начало XX века) стояли на позициях химической природы катализа, только в конце 50-х годов XX века стало окончательно ясно, что катализ химическое явление[3] .
Идеальный катализатор – это химическое соединение или простое вещество, которое ускоряет одну из термодинамически возможных реакций и не участвует в стехиометрическом уравнении этой реакции. Ускорение реакции происходит в результате образования промежуточных соединений и появления нового, более выгодного пути на поверхности потенциальной энергии (нового механизма).
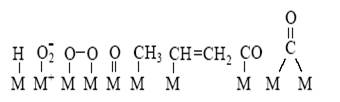
Если обозначить катализатор буквой K (комплекс металла, молекула кислоты, активный центр на поверхности, молекула фермента), то простейшим механизмом каталитической реакции,
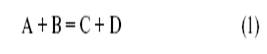
будет двухстадийный механизм
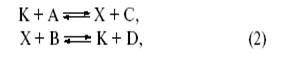
где X – промежуточное соединение.
Таким образом, каталитический процесс – это совокупность обычных химических реакций (в растворе, на поверхности или в газе), но совокупность особенная, имеющая циклический характер.
Циклическую природу каталитического процесса можно наглядно представить в виде графа, у которого в вершинах (кружках) будут находиться
промежуточные вещества и катализатор, а линии, связывающие вершины (ребра), будут соответствовать стадиям механизма. Тогда схема (2) будет представлена простым циклическим графом (рис. 1).
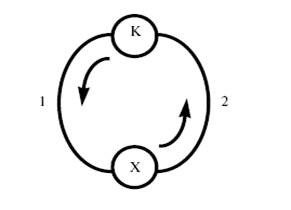
Рис. 1. Граф механизма (2) каталитической реакции.
Влияние различных факторов на каталитическую реакцию (особенности электронного строения твердого тела и его поверхности, геометрия поверхности, особенности электронного строения комплексов металлов, свойства растворителей и др.) не отличается от влияния тех же факторов на любую химическую реакцию.
Рассмотрим историю открытия и механизм очень интереснойс химической точки зрения и промышленно важной реакции[4] . В 1894г. Ф. Филлипс заметил, что этилен и СО восстанавливают влажный PdCl2 до металлического палладия. В продуктах окисления этилена Филлипс качественно обнаружил ацетальдегид. В конце 30-х годов, изучая действие воды на комплексы PdCl2 с этиленом, также наблюдали быстрое восстановление PdCl2 и образование ацетальдегида. Эта удивительная реакция не заинтересовала химиков-органиков, поскольку реакция
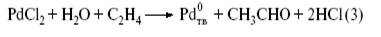
– стехиометрический синтез ацетальдегида из этилена и палладия(II).
Господствовавшие в тот период представления о том, что каталитический процесс не может состоять из стадий образования и превращения обычных химических соединений и веществ, не позволили сделать следующий логичный шаг – окислить до PdCl2 в том же реакторе, хотя такие реакции химикам были уже известны. Добавим в раствор PdCl2 хлорид меди(II):
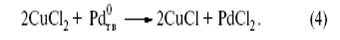
Сложив уравнения (3) и (4), получим
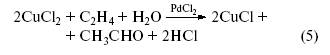
– процесс окисления этилена хлоридом меди(II), катализируемый PdCl2.
Очень просто и этот стехиометрический по CuCl2 процесс сделать каталитическим. Давно известно, что CuCl легко окисляется кислородом в слабокислых растворах:
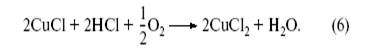
В результате (3) + (4) + (6) получаем изящную каталитическую реакцию
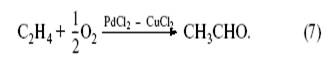
Это все и было проделано двумя группами исследователей – группой Ю. Смидта и группой химиков в Москве – И.И. Моисеевым, М.Н. Варгафтиком и Я.К. Сыркиным[5] . Сейчас эта реакция (промышленное название Вакер-процесс[6] ) является лучшим промышленным методом получения ацетальдегида.
Как мы видим, каталитический процесс включает совокупность обычных химических реакций, организованных так, что часть реагентов (PdCl2 , CuCl2 ) регенерируется в стадиях процесса и не входит в стехиометрию итоговой реакции. Хлорид палладия в этой реакции ускоряет процесс присоединения ОН-группы (из молекулы воды) к этилену.
В образующемся металлоорганическом соединении ClPdCH2 CH2 OH Pd(II) окисляет связанную с ним органическую группу CH2 CH2 OH до ацетальдегида.
Мы остановились подробно на этой реакции, поскольку она наглядно демонстрирует и химическую природу катализа, и основные принципы действия катализаторов.
По условиям проведения каталитические процессы бывают гомогенными (реакция протекает в объеме раствора или в объеме газовой фазы) и гетерогенными (реакция идет на поверхности твердого тела). При наличии двух фаз (жидкость–жидкость, жидкость–твердое тело) используют также катализаторы – переносчики реагентов из одной фазы в другую (межфазный катализ) (рис. 2).
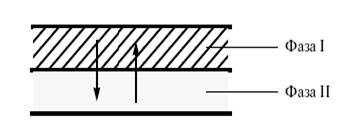
Рис. 2. Катализ межфазного переноса (межфазный катализ).
При этом катализаторы межфазного переноса выполняют не только физическую (транспортную) функцию, но и существенно влияют на реакционную способность переносимой частицы[7] .
Среди множества требований к промышленным катализаторам следует отметить три главных:
1) каталитическая реакция должна протекать с заметной скоростью (активность катализатора);
2) скорость основной реакции должна существенно превышать скорости всех остальных реакций (селективность действия катализатора);
3) активность катализатора не должна заметно снижаться во времени (стабильность работы катализатора, время его жизни).
Активность катализатора прежде всего характеризуется скоростью каталитической реакции r (моль/л с). Для сравнения активности различных катализаторов используют величину A , называемую частотой оборотов катализатора. Эту величину обычно получают как отношение начальной, или
стационарной, скорости реакции r к начальной концентрации катализатора C 0 (моль/л):
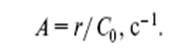
Строго говоря, для определения А надо знать не общее количество загруженного катализатора, а число молей активной в катализе формы катализатора или количество молей активных центров на единице поверхности или в единице объема твердого катализатора. Величина 1/А характеризует время одного оборота, т.е. одного каталитического цикла (см. рис. 1). Очевидно, что чем больше величина А , тем меньшее количество катализатора можно использовать в процессе. Величина А меняется в случае химических катализаторов в очень широком диапазоне от 10 −3 до 104 с−1 .
В ферментативных процессах А достигает 106 с −1 . Очевидно, что очень важно, сколько часов будет работать катализатор с активностью А .
Время жизни промышленных катализаторов колеблется от нескольких часов до 2–3 лет. Чем выше величина А , тем меньше может быть допустимое время жизни катализатора.
Как мы видим, в отличие от идеального катализатора, который, не участвуя в стехиометрии реакции, не расходуется, реальный катализатор может дезактивироваться (отравление каталитическими ядами, закоксовывание и др.) и тогда на 1 кг продукта будет расходоваться какое-то количество катализатора.
Очень важной характеристикой катализатора является селективность S катализируемой им реакции, поскольку именно селективность определяет непроизводительные затраты сырья и энергии на выделение продукта и переработку отходов.
2. ОСНОВНЫЕ ТИПЫ КАТАЛИЗАТОРОВ И МЕХАНИЗМЫ КАТАЛИТИЧЕСКИХ РЕАКЦИЙ.
Практически все катализаторы можно разделить на 5 типов, учитывая особенности их строения и механизма катализа[8] .
1. Кислоты и основания (гомогенные и гетерогенные катализаторы) – протонные кислоты Бренстеда (НА) в водных и неводных средах, апротонные кислоты Льюиса–Усановича (BF3 , RI), протонные и апротонные центры твердых оксидов (γ-Al2 O3 ,Al2O3 –SiO2 , цеолиты), любые типы оснований (в том числе твердые – MgO, CaCO3 , анионообменные смолы).
2.Комплексы металлов (гомогенные и гетерогенные катализаторы) – MLn, MmLn.
3.Твердые соединения металлов типа MmЭn, где Э = O, S, Se, Te, As, P, C, N, Si, B, H, – гетерогенные катализаторы.
4.Металлические катализаторы (гетерогенные) – нанесенные на инертных носителях (Pt/Al2 O3 ) или массивные металлы и сплавы.
5.Ферменты (гомогенные и гетерогенные).
Рассмотрим особенности механизма действия этих групп катализаторов.
Кислотно-основной катализ относится к очень распространенному и к наиболее изученному типу катализа. В катализе протонными кислотами Бренстеда (НА) субстрат реакции (реагент) выступает в качестве основания и первой стадией является протонирование реагента. Протонированный реагент (B) переходит в более реакционно-способное состояние и превращается далее через одно или несколько промежуточных соединений.
Например, механизм превращений олефинов в присутствии кислоты НА может быть представлен схемой 1.
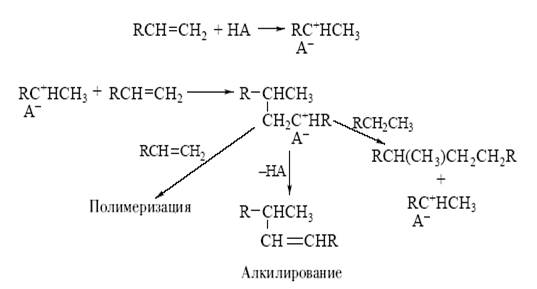
Схема 1.
В результате первой стадии переноса протона на олефин образуется новая кислота (апротонная) – ион карбения. Эта частица содержит положительно заряженный атом углерода (карбокатион) с вакантной орбиталью:
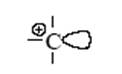
Такой катион (R+ – кислота по Льюису) реагирует со второй молекулой олефина как с основанием и вновь образует ион карбения. Этот новый катион может отщепить кислоту-катализатор, и тогда мы получим продукт каталитической димеризации олефина. Этот же катион может прореагировать последовательно с несколькими молекулами олефина, что приведет к процессу полимеризации олефина.
Если в системе присутствует соответствующий алкан, то ион карбения, отщепляя от него гидридион (H− ), превратится в изопарафин, а из алкана образуется новый ион карбения. В этом случае мы получим продукт алкилирования парафина олефином, причем образовавшийся на первой стадии ион карбения и будет катализатором процесса алкилирования.
Любое органическое и неорганическое соединение может выступать в роли основания, однако чем слабее основность соединения, тем более сильная кислота требуется для его протонирования. Так, очень сильные протонные кислоты (“суперкислоты”, “магические” кислоты), образующиеся в системах HF–SbF5 , ( H + [ SbF -6 ]), и HSO3 F–SbF5 (H2 SO 3 F + [ SbF 5 ( OSO 2 F )− ]) протонируют в мягких условиях даже парафины[9] . Так, метан образует ион карбония (ион метония).
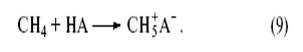
По аналогии с трехцентровыми двухэлектронными связями в диборанах (B2 H6 ) или в Al2 (CH3 )6 строение иона метония CH+ 5 можно представить структурой
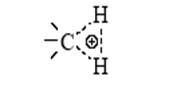
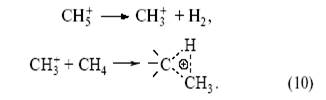
(два электрона С–Н-связи обслуживают три центра). Образующаяся частица CH+ 5 может отщепить Н+ (образуется метан) или CH+ 3 (образуется H2 ). Ион карбения CH+ 3 реагирует по тому же механизму с молекулой CH4 , что и H+ .
При этом образуется этан. В результате из метана (в мягких условиях) получаются парафины C 2 , C3 и C4 :
За исследования карбокатионов в растворах “суперкислот” Дж. Ола получил Нобелевскую премию. На поверхности ряда оксидов (γ-Al2 O3 – алюмосиликаты) присутствуют протонные и апротонные кислотные центры[10] . При этом сила протонных центров ряда алюмосиликатов может приближаться к силе концентрированной серной кислоты. Особенно интересный тип кристаллических алюмосиликатов (цеолитов) широко применяется в промышленном катализе.
Металлокомплексный катализ – быстроразвивающаяся область каталитической химии. Более 50 крупнотоннажных промышленных процессов используют гомогенные или гетерогенные металлокомплексные катализаторы. Химия комплексных соединений (координационная химия) и химия металлоорганических соединений являются основой этой области каталитической химии.
Координационная химия после создания теории комплексных соединений А. Вернером (1893 – 1905гг.) прошла большой путь и стала, по существу, языком неорганической и металлоорганической химии. Установлено, что простых соединений, в которых число двухэлектронных связей соответствует степени окисления металла комплексообразователя, практически не существует.
Так, например, молекула HgCl2 существует в виде линейной молекулы Cl–Hg–Cl только в парах при высоких температурах (>100°С). В твердой фазе и в растворах как соли, так и гидроксиды тяжелых металлов существуют в виде координационных соединений, в которых атом металла окружен различными группами (лигандами), например октаэдр HgCl2 (H2 O)4 в воде.
Мы рассмотрим лишь несколько примеров типичных комплексов металлов, чтобы продемонстрировать разнообразие лигандов (атомов, фрагментов молекул и молекул) и показать, что фактически любая молекула или частица (то есть любой участник каталитической реакции) может находиться в координационной сфере металла.
В табл. 1 приведены нейтральные молекулы и анионы, которые могут служить донорами σ- и π- электронов при образовании координационных связей M–L.
Таблица 1
Типы легандов в комплексных соединениях

K[PtCl3 (C2H4 )] – первое соединение, описанное в литературе, содержащее молекулу этилена, связанную с атомом металла за счет пары π-электронов этилена (соль Цейзе). Комплексы металлов с органическими лигандами CH+ 5 и с производными бензола (аренами) были синтезированы в 1951 – 1955 гг. Их структура была весьма необычна для того времени: органические ароматические лиганды оказались связаны как π-лиганды с центральными атомами металла.
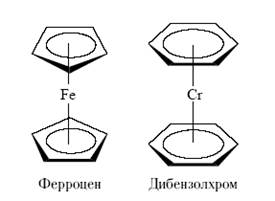
За вклад в развитие этой области химии Дж. Уилкинсон и Э.О. Фишер получили Нобелевскую премию.
Степень окисления металла в комплексах может быть положительной (PdCl2- 4 , Ni (H2 O)2+ 6, HPtCl(PR3 )2 , K2 ReH9 , K3 W(CH3 )6 ), равняться нулю (Ni (CN4- 4 ), Ni(CO)4 , Cr(C6 H6 )2 ) или даже быть отрицательной (Na2 Fe(CO)4 ).
К фундаментальным открытиям последнего времени следует отнести синтезы стабильных комплексов с молекулярным водородом (Г. Кубас, 1984), в которых молекула Н–Н связана с металлом за счет своей пары электронов (без разрыва связи Н–Н) (аналогично связи CH+ 3 c H2 ): W(CO)3 (PR3 )2 (H2 ), Ir (H)2 (H2 )2 PR3 + 2.
Помимо комплексов с одним центральным атомом металла известны комплексы состава MmLn. Это комплексы с мостиковыми лигандами.
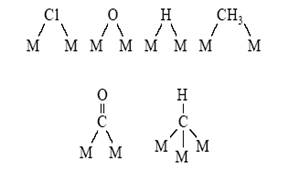
и комплексы со связями М–М (кластеры металлов). Связи металл–металл в кластерах имеют различную кратность[11] :
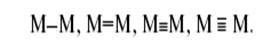
Двойные и тройные связи близки по свойствам к π-связям в органических (C2 H4 , C2 H2 ) и неорганических (N2 ) молекулах. В отличие от иона H+ ионы переходных металлов могут координировать до 9 лигандов, то есть связывать несколько молекул и фрагментов молекул в координационной сфере металла. При этом атом или ион переходного металла может быть не только акцептором пары электронов  , но и донором неподеленных пар d -электронов на лиганд , но и донором неподеленных пар d -электронов на лиганд  что очень важно для активации координированной частицы. Активация различных молекул особенно эффективно осуществляется в кластерных комплексах[12] . что очень важно для активации координированной частицы. Активация различных молекул особенно эффективно осуществляется в кластерных комплексах[12] .
Рассмотрим механизм реакции карбонилирования метанола, которая является сегодня лучшим методом получения уксусной кислоты.
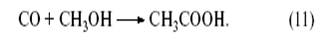
В этом процессе действуют два катализатора – металлокомплексный Rh(I) и кислотный (HI). Метанол реагирует с HI с образованием CH3 I и H2 O:
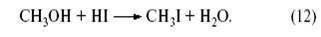
Иодистый метил присоединяется к комплексному аниону Rh (CO2 )I- 2 с образованием комплекса Rh(III) со связью CH3 −Rh:
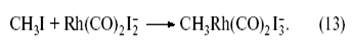
В полученном комплексе Rh(III) мы видим группу CH3 и координированные родием молекулы СО. Далее происходит замечательная внутрисферная реакция объединения двух групп (CH3 и CO) – реакция внедрения СО по связи CH3 –Rh. Атакующая комплекс Rh(III) молекула СО занимает координационное место, освободившееся после связывания СО группой CH3 . Образующийся ацетильный фрагмент (CH3 CO) остается связанным родием(III):
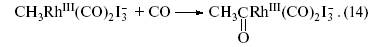
Затем происходит внутрисферная реакция объединения ацетильной группы с атомом иода (восстановительное элиминирование) с возвращением исходной формы катализатора и гидролиз иодангидрида уксусной кислоты с образованием HI и CH3 COOH:
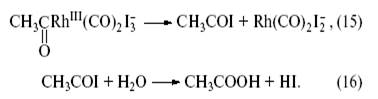
Таким образом, Rh(I) катализирует карбонилирование CH3 I до CH3 COI, а HI – карбонилирование метанола до уксусной кислоты.
Роль металлоорганических соединений в катализе особенно оценили после открытия гомогенных и гетерогенных катализаторов для стереоспецифической полимеризации олефинов и диенов – TiI4 –Al(C2 H5 )3 , TiCl3 –Al(C2 H5 )3 . Авторы этих каталитических систем К. Циглер и Дж. Натта.
Важнейшую роль в развитии металлокомплексного органического катализа сыграло открытие реакций окисления олефинов в растворах солей палладия, о котором мы говорили выше, и изучение их механизма (И.И. Моисеев, П. Генри).
Гетерогенный катализ металлами и оксидами металлов. Каталитические реакции на поверхности так же, как и реакции на поверхности электродов (электрохимия) и фотохимические процессы в тонких пленках (фотография), относятся к особой области химии – химии поверхности.
В 50-е годы процессы адсорбции различных веществ на поверхности (первая химическая стадия в гетерогенном катализе) стали изучать на молекулярном уровне многочисленными физическими методами[13] . К реакциям на поверхности переходных металлов, оксидов металлов и других металлсодержащих соединений применимы все представления координационной и металлоорганической (в случае органических реакций) химии[14] . На поверхности металлов образуются первичные комплексы реагентов с атомом или группой атомов поверхности и продукты их превращений.
Например, при адсорбции этилена на поверхности кристаллического Rh происходят следующие химические реакции при изменении температуры (Г. Саморджай) (схема 2):
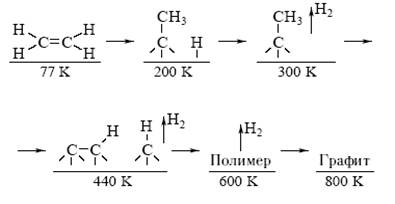
Схема 2
Адсорбированные молекулы изменяются так же сильно, как и в процессах комплексообразования (особенно с кластерами металлов). Например, молекула бензола (средняя длина С–С = 1.40 A), взаимодействуя с 4 атомами Rh на поверхности, растягивается так, что длины двух противоположных С–С-связей становятся равными 1.63 A (больше простой С–С-связи в этане), а длины 4 других С–С-связей равны 1.45 A.
Помимо химических проявлений, в гетерогенном катализе следует учитывать наличие объема твердого тела (большого количества атомов) и ряд особенностей поверхности[15] .
1. Поверхность с макроскопической точки зрения является объектом с очень сложным рельефом, а твердое тело обычно имеет развитую пористую структуру. Это очень хорошо демонстрирует электронно-микроскопическая фотография поверхности активированного угля (типичный катализатор или носитель в гетерогенном катализе) (рис. 3).
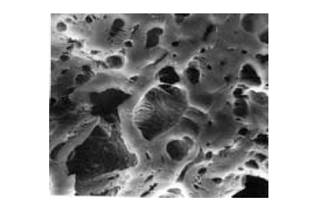
Рис. 3. Электронно-микроскопическая фотография поверхности активированного угля
Даже на поверхности монокристаллов металлов существуют площадки, выступы, ступеньки и трещины (рис. 4). Это сказывается на геометрии окружения различных атомов и, естественно, на их реакционной способности.
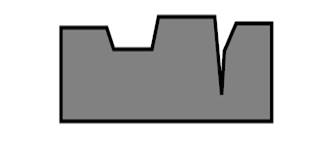
Рис. 4. Геометрия поверхности монокристалла.
2. В случае металлического катализатора атом металла (или группа атомов) на поверхности реагирует как локальный активный центр с молекулой реагента, однако этот атом окружен другими атомами (как лигандами), которые меняют свойства реагирующего атома. Так же влияют атомы кислорода на свойства металла в оксиде.
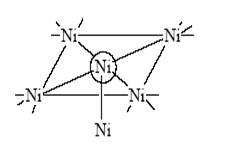
3. В решетке оксида металла присутствуют различные дефекты, примеси, что приводит к появлению небольшого количества ионов металла в более низких или в более высоких степенях окисления (в объеме и на поверхности). Например, в оксиде хрома (Cr2 O3 ) могут присутствовать ионы Cr2+ (лишние электроны) и ионы Cr4+, Cr5+ , Cr6+ (электронные вакансии).
При этом реагент (молекула углеводорода, например) реагирует с Cr2+ и Cr4+ поразному, образуя гидрид-ион в первом случае (Cr3+ –H– ) и протон во втором (Cr3+ –OH+ ) и радикал R (свободный или связанный с ионами хрома).
4. Свойства поверхностных соединений (двухмерный адсорбционный слой) отличаются от свойств тех же соединений, образующих обычную (трехмерную) фазу.
5. Объемная фаза твердого тела в ряде случаев также участвует в каталитическом акте. Происходит диффузия атомов из решетки к поверхности (Н, О), а также возможен перенос электронов через объемную фазу от одного центра поверхности к другому.
6. Роль локальных свойств активного центра и роль коллективных свойств твердого тела определяется типом катализатора и механизмом реакции.
7. В случае смешанных оксидов металлов или сплавов металлов концентрации компонентов в объеме и на поверхности различаются: поверхностный монослой атомов может заметно обогащаться одним из элементов (металлов).
Простейший механизм реакции окисления СО на поверхности оксида цинка описывается схемой 3.
Вначале происходит образование поверхностных карбонатов, распад которых ускоряется адсорбирующимся кислородом [12]:
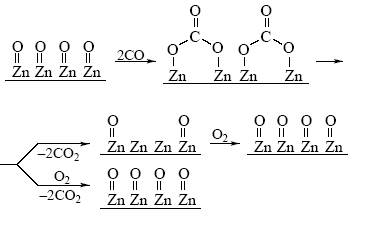
Схема 3
Биокатализаторы – ферменты. Ферменты – это молекулы белка, которые в большинстве случаев растворимы в воде, но иногда находятся в коллоидном (микрогетерогенном) состоянии. Активные центры фермента формируются в результате стягивания в одну область пространства различных функциональных групп, принадлежащих различным аминокислотным фрагментам молекулы белка (COOH,
OH,
NH2,
SH,
имидазол и др.).
Активный центр располагается в виде щели в глобуле белковой молекулы. В некоторых ферментах и коферментах (молекулах, выполняющих роль специфических реагентов, регенерируемых в ходе реакций с другими ферментами) присутствуют ионы металлов (Fe, Cu, Zn, Mo, V, Co и др.), окруженные макролигандами.
Одним из таких Co-содержащих коферментов является витамин В12 . Наряду с высокой частотой оборотов фермента (А) при относительно небольшом времени жизни ферментам свойственна высокая специфичность по отношению к определенному субстрату и высокая специфичность к типу реакции.
Высокие активность и селективность действия фермента достигаются благодаря высокой степени организации активного центра и некоторым особенностям действия белковых катализаторов:
а) первичное связывание реагента в активном центре весьма специфично;
б) молекула реагента, попав в активный центр, вызывает изменение структуры белковой молекулы и подстраивает “под себя” геометрию активного центра так, чтобы дальнейшее превращение было наиболее выгодным (протекало с большей скоростью);
в) в активном центре на молекулу реагента обычно синхронно действуют несколько активных групп (кислотных, основных, нуклеофильных каталитических центров), что напоминает действие сварочных роботов на конвейере сборки автомобилей.
Роль объединения различных групп в одном катализаторе и синхронного их действия давно известна и в химическом кислотно-основном катализе.
3. МЕСТО КАТАЛИТИЧЕСКОЙ ХИМИИ В СИСТЕМЕ ХИМИЧЕСКИХ ЗНАНИЙ.
В первой половине ХХ века две области химии – неорганическая и органическая – развивались весьма обособленно. Сильно различались структуры соединений, использовались разные подходы к реакционной способности, нарабатывались свои собственные эмпирические правила[16] .
В неорганической химии активно синтезировали и изучали комплексные соединения с центральным атомом металла (особенно в растворах), изучали структуры и свойства кристаллических и аморфных соединений металлов (оксиды, соли, карбиды, нитриды, гидриды) и интерметаллидов.
Связь элемент–элемент была характерной для бороводородов, полимеров серы (S8, S12) и фосфора (P4), твердых металлов и интерметаллидов (InAs, Nb3Sn, Fe3Ni, LaNi5 и др.)
Химики-органики имели дело в основном с линейными и циклическими цепочками из атомов углерода и с соединениями, содержащими связи элемент–углерод (элемент – O, Hal, N, S, Si, P, B, Al, щелочные и щелочно-земельные и непереходные металлы). Общими для этих двух областей химии
были лишь представления о локализованных двухэлектронных двухцентровых связях и метод валентных схем (метод резонанса), хотя в неорганической химии отдавали предпочтение теории кристаллического поля (для комплексов переходных металлов).
Начиная с открытия структур ферроцена и дибензолхрома быстрыми темпами стала развиваться химия металлоорганических соединений переходных металлов, особенно после того как были подвергнуты ревизии правила Менделеева и Несмеянова о принципиальной нестабильности связи М–С, если М – переходный металл[17] .
Стало ясно, что дело не в прочности связи (энергия связи Pt–C оказалась в 2 раза и более выше энергии связи Hg–C в стабильных ртутьорганических соединениях), а в кинетической лабильности таких молекул, то есть в их очень высокой реакционной способности[18] .
Вторая половина текущего века ознаменовалась синтезом соединений с цепочками и циклами из атомов металла, связанных с лигандами, в неорганической и металлоорганической химии (кластеры металлов):
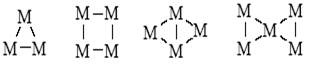
В органической химии синтезировали каркасные соединения (полиэдраны) – призман, тетраэдран, кубан и др. Такие же и более сложные полиэдры, но из атомов металла получены в координационной химии:
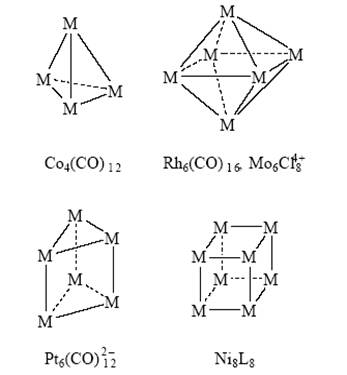
Рекордные значения m в металлических кластерах получены в комплексах палладия (m = 561)[19] .
К началу 1990-х годов стало ясно, что нет принципиальной разницы в структурах молекул, содержащих скелеты из атомов металла, атомов углерода или из атомов других элементов. Химия самого углерода пополнилась новым классом молекул – полиэдранов Сn(С60 , С70 и др.).
Молекула С60 (фуллерен), растворимая в ароматических углеводородах, имеет структуру усеченного икосаэдра и участвует в разнообразных химических реакциях.
Метод молекулярных орбиталей становится единым подходом для описания природы связи в любых соединениях и их реакционной способности. Предложенный Р. Хофманом анализ фрагментов и “принцип изолобальности” завершил объединение теоретических подходов к реакционной способности органических, металлоорганических и металлокомплексных соединений[20] .
Согласно этому принципу, легко реагируют группы (фрагменты молекул, молекулы), имеющие одинаковое число электронов на граничных орбиталях, одинаковую симметрию орбиталей и близкие энергии орбиталей (изолобальные группы).
Так, группы :СH2 и :Fe(CO)4 изолобальны и поэтому могут образовать следующий ряд соединений:
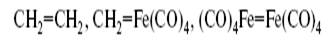
(Fe2 (CO)8 присоединяет еще одну молекулу СО и дает стабильный Fe2 (CO)9 ):
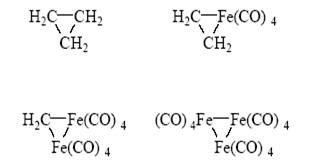
Таким образом, появились единые типы структур и единый язык теоретического анализа. Естественно, каждая область химии продолжает развиваться в рамках своих собственных задач и внутренней логики, однако можно утверждать, что концептуальное объединение в химии (структуры, природа связи, теория реакционной способности) совершилось.
На фоне объединения различных областей химии и продолжающейся дифференциации химических наук (биоорганическая и бионеорганическая химия) рассмотрим эволюцию химии каталитических процессов. Из скромного раздела физической химии (химическая кинетика и катализ) действительно сформировалась самостоятельная область химии – каталитическая химия.
Интересно отметить, что еще в пятидесятые годы А.А. Баландин часто использовал такое название, считая каталитическую химию вершиной химических знаний[21] .
Сегодня каталитическая химия и концептуально, и предметно, на уровне веществ, объединила различные области химии.
Во-первых, каталитическая химия вобрала в себя все достижения координационной, органической и металлоорганической химии, ферментативного катализа, химии твердого тела, теории растворов и теории реакционной способности.
Во-вторых, исходный катализатор (активный центр) и все промежуточные вещества в катализе металлсодержащими катализаторами практически всегда суть координационные соединения, а в катализе органических реакций промежуточные вещества почти всегда являются и металлоорганическими соединениями. Если еще принять во внимание сложность организации каталитического процесса (циклический характер), можно заключить, что каталитическая химия представляет собой сегодня высшую ступень эволюции химических знаний.
Заключение
Итак, катализ – это ускорение или возбуждение химических реакций в присутствии веществ – катализаторов, многократно вступающих в промежуточное химическое взаимодействие с участниками реакции и восстанавливающих после каждого цикла свой первоначальный химический состав[22] . Это определение не содержит никаких указаний на причину, по которой такое промежуточное взаимодействие между катализатором и реагентами вызывает ускорение химической реакции, но лишь описывает новый путь, по которому идет эта реакция с участием катализатора.
Катализ занимает особое место как в системе наших знаний о веществах и их превращениях, так и в практической деятельности человека. Он лежит в основе существования растительного и животного мира, обеспечивая с помощью ферментов функционирование живых систем. В истории нашей цивилизации катализ не раз становился решающим фактором технического прогресса. Достаточно назвать лишь три промышленных каталитических процесса: синтез аммиака, крекинг нефти и полимеризация олефинов, чтобы убедиться в этом.
Все сказанное в достаточной мере характеризует ту важную роль, которую играет катализ в сфере научной и практической деятельности современного человека. Существует, однако, одна специфическая особенность катализа, которая делает постановку вопроса, вынесенного в заголовок статьи, хотя и несколько неожиданной, но вполне оправданной. В отличие от других разделов химии в катализе пока не существует общей теории, способной заранее предсказать, будет ли данное вещество ускорять какую-либо реакцию.
Широко известные теории катализа такие, как мультиплетная теория Баландина, теория активных ансамблей Кобозева, теория катализа на полупроводниках Доудена и многие другие теоретические концепции являются не общими, а частными моделями, относящимися к сравнительно узкому кругу каталитических систем. Но и эти частные теории лишь объясняют опытные факты.
Отсутствие научной теории, способной предсказать каталитические свойства веществ, как раз и породило неоднократно встречавшееся в специальной литературе суждение о катализе скорее как об искусстве, чем науке. Полуторавековая история катализа как самостоятельного раздела химии показывает, что все известные катализаторы были открыты либо случайно, либо интуитивно, эмпирически.
Объективную причину столь необычной в современной науке ситуации следует, по-видимому, искать в самой природе катализа и поразительном разнообразии каталитических процессов. Даже сейчас, когда новейшие экспериментальные методы позволяют подойти к изучению процессов катализа, а также самих катализаторов на атомно-молекулярном уровне и в масштабе реального времени, Природа не открывает тайну, как она это делает.
Как в свое время отмечал Н.Н. Семенов, вопрос о теории катализа лежит в контексте более общей проблемы реакционной способности веществ. Принципиальные пути решения этой проблемы квантово-химическими методами сейчас достаточно детально разработаны, и прогресс полностью определяется уровнем развития вычислительной техники.
Список использованной литературы
1. Боресков Г.К. Катализ. Избранные труды. – Новосибирск: Наука, 2004.
2. Боресков Г.К. Некоторые проблемы катализа. – М.:Знание, 2001.
3. Волков В.А., Вонский Е.В., Кузнецов Г.И. Выдающиеся химики мира. Биографический справочник. / Под ред. В.И.Кузнецова. М.: Высшая школа, 2004.
4. Гейтс Б., Кетцир Дж., Шуйт Г. Химия каталитических процессов. – М.: Мир, 2000.
5. Егоров В.В. Теоретические основы неорганической химии. Краткий курс для студ.cельхоз.вузов: Учебник. – Спб.: Лагь. – 2005. – 192 с. |
 Скачать 230.67 Kb.
Скачать 230.67 Kb.