фенолы. Лекция 24 фенолы
 Скачать 1.82 Mb. Скачать 1.82 Mb.
|

|
ЛЕКЦИЯ 24 ФЕНОЛЫ 1. Классификация. Изомерия. Номенклатура. Многоатомные фенолы. Основные пути использования фенолов. 2. Способы введения гидроксильной группы в ароматическое ядро: щелочное плавление солей сульфокислот, гидролиз галогенпроизводных, синтез с использованием солей диазония, кумоловый способ получения фенола. 3. Химические свойства. Причины повышенной кислотности фенолов по сравнению с алифатическими спиртами, влияние заместителей на кислотность фенолов. Получение фенолятов простых и сложных эфиров. Реакции электрофильного замещения: галогенирование, сульфирование, нитрование. Фенолами называются производные аренов, в которых один или несколько атомов водорода ароматического кольца замещены на гидроксильные группы. 24.1. Классификация. Изомерия. Номенклатура. Многоатомные фенолы. Основные пути использования фенолов Классификация и номенклатура. По количеству ароматических ядер в молекуле различают собственно фенолы, а также нафтолы, антролы, фенантролы и др.: 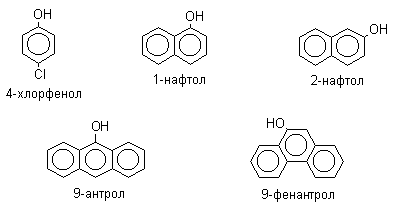 Рис. 24.1. Классификация фенолов по числу ароматических ядер в молекуле По числу гидроксильных групп различают одно-, двух-, трех-, многоатомные фенолы: 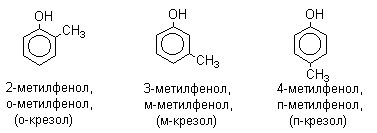 Рис. 24.2. Классификация фенолов по числу гидроксильных групп в молекуле: одноатомные фенолы 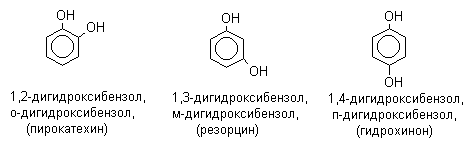 Рис. 24.3. Классификация фенолов по числу гидроксильных групп в молекуле: двухатомные фенолы 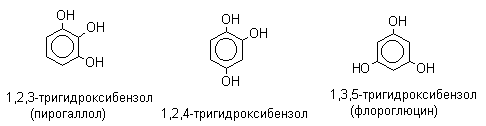 Рис. 24.4. Классификация фенолов по числу гидроксильных групп в молекуле: трехатомные фенолы 1. При построении названий фенолов атомы углерода в бензольном кольце принято обозначать от 1 до 6, начиная с углерода, связанного с ОН-группой (номенклатура схожа с ароматическими углеводородами). 2. Цифрами и приставками указывают положение и число заместителей, с добавлением основы – фенол. 3. Простейший фенол – гидроксибензол С6Н5ОН называют просто фенол. 4. При наличии нескольких заместителей начало нумерации определяет гидроксильная группа и эти соединения рассматриваются как производные фенола. 5. Иногда в соединениях сложного строения наличие гидроксильной группы обозначают префиксом гидрокси-. 6. Многие фенолы имеют тривиальные названия, принятые номенклатурой ИЮПАК (см. рис. 24.1-24.4). Изомерия. Как видно из приведенных примеров, фенолам свойственна структурная изомерия (изомерия положения гидроксигруппы). Использование фенолов. На основе фенола получают: - лекарственные препараты, например, аспирин, салол, фенолфталеин; - красители; - парфюмерные продукты; - пластификаторы для полимеров; - средства защиты растений. Раствор фенола используют в качестве дезинфицирующего средства (карболовая кислота). Двухатомные фенолы – пирокатехин, резорцин, гидрохинон (пара-дигидроксибензол): применяют как антисептики (антибактериальные обеззараживающие вещества); входят в состав дубителей для кожи и меха; используют как стабилизаторы смазочных масел и резины; используют для обработки фотоматериалов и как реагенты в аналитической химии. В виде отдельных соединений фенолы используются ограниченно, зато их различные производные применяют широко. Фенолы служат исходными соединениями для получения разнообразных полимерных продуктов – феноло-альдегидных смол, полиамидов, полиэпоксидов. Физические свойства. Фенол и его низшие гомологи представляю собой бесцветные низкоплавкие кристаллические вещества или жидкости с сильным характерным запахом. Двух- и трехатомные фенолы - твердые вещества с более высокими температурами плавления, без запаха. Фенолы малорастворимы в воде, но легко растворяются в неполярных растворителях: эфире, бензоле, спирте и хлороформе. Способы получения. Основными способами получения фенолов являются синтетические способы, основанные на введении гидроксильной группы опосредованно вместо имеющихся в бензольном кольце заместителей, например путем замещения атома хлора. Гидролиз галогенпроизводных. В промышленности используют способ взаимодействия хлорбензола с раствором щелочи NaOH, при высоких температурах (в зависимости от особенностей технологического регламента, от 280ºС до 350ºС) и высоком давлении (порядка 30 МПа): Рис. 24.5. Схема реакции гидролиза галогенпроизводных бензола Реакция идет в две стадии, сначала получение фенолята натрия, затем его реакция с соляной кислотой: Рис. 24.6. Схема реакции получения фенола гидролизом хлорбензола Щелочное плавление солей ароматических сульфоновых кислот. Способ заключается в сплавлении натриевых и калиевых солей ароматических сульфоновых кислот с твердыми щелочами при температуре 300-350ºС. Примером может служить синтез β-нафтола из нафталина: 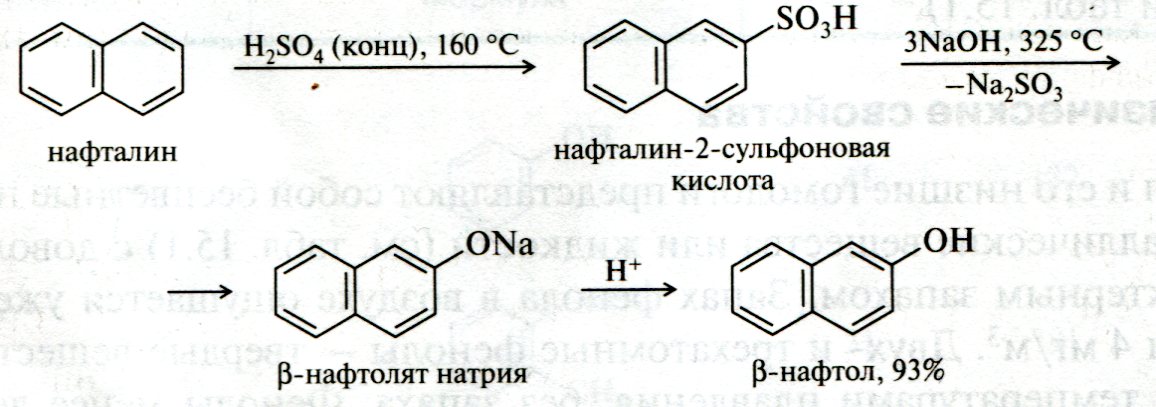 Рис. 24.7. Схема получения β-нафтола из нафталина 1. Образующуюся на первой стадии нафталин-2-сульфоновую кислоту переводят в натриевую соль, которую сплавляют со щелочью, поэтому данный способ называют щелочным плавом. 2. После подкисления минеральной кислотой выделяется конечный продукт - β-нафтол. Многатомные фенолы тоже могут быть получены способом щелочного плава. Так, для получения пирокатехина используют о- или п-гидроксибензолсульфоновые кислоты, для синтеза резорцина – бензол-1,3-дисульфоновая кислота: 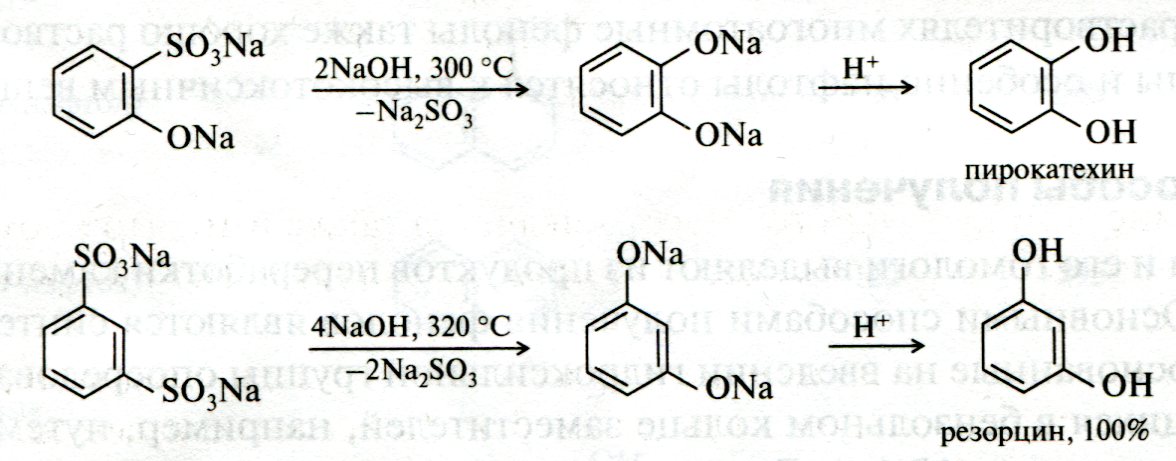 Рис. 24.8. Схема получения двухатомных спиртов: пирокатехина и резорцина Основным недостатком способа являются жесткие условия процесса и большое количество отходов, загрязняющих окружающую среду. Кумольный способ (окисление изопропиларенов, изпропилбензол - кумол). Преимущество метода: безотходная технология (выход полезных продуктов > 99%).В настоящее время кумольный метод используется как основной в мировом производстве кумола (до 80% всего производства). Кумольный способ включает в себя две последовательные стадии. 1. Сначала кумол окисляют кислородом воздуха в гидропироксид кумола. Это радикальное окисление протекает по третичному атому углерода с образованием промежуточного устойчивого радикала бензильного типа. 2. На второй стадии достаточно стабильный гидропироксид кумола разлагают разбавленной серной кислотой: 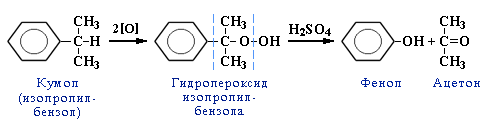 Рис. 24.9. Схема получения фенола окислением изопропилбензола Достоинством кумольного способа является также и то, что кроме фенола получается ацетон (на 1 кг фенола образуется 0,6 кг ацетона). Аналогичным путем из м- и п-цимолов (м- и п-метилкумолов) получают м- и п-крезолы, из 2-изопропилнафталина получают β-нафтол. Разложение солей арилдиазония. Этот важный лабораторный способ синтеза фенолов заключается в переводе ароматической группы в диазогруппу с последующим замещением диазогруппы на гидроксильную: 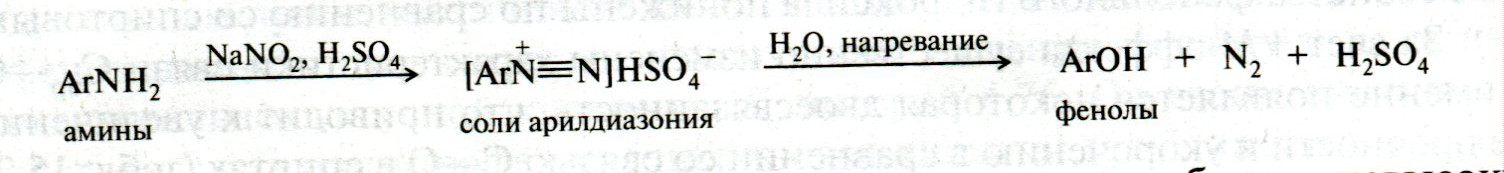 Рис. 24.10. Схема получения фенола разложением солей диазония Выходы фенолов при разложении солей арилдиазония обычно невысоки (50-60%). Этот способ использую в случае: - необходимо получить замещенный фенол, свободный от изомеров; - иные способы получения фенолов менее эффективны. Химические свойства. Реакционными центрами в молекуле фенолов являются фенольная гидроксильная группа и ароматическая группа, которые взаимно влияют друг на друга: 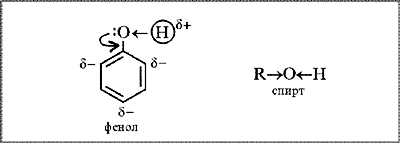 Рис. 24.11. Электронная формула фенола Причины повышенной кислотности фенолов по сравнению со спиртами. Фенольная гидроксильная группа за счет р,π-сопряжения с ароматическим кольцом обусловливает дефицит электронной плотности на атоме кислорода, в результате чего протон гидроксильной группы становится более подвижным, чем в спиртах, т.е. увеличивает кислотные свойства. Благодаря электронодонорному влиянию фенольного гидроксила (+М-эффект ОН-группы) электронная плотность ароматического кольца повышена по сравнению с бензолом, особенно в орто- и пара-положениях. Это существенно облегчает протекание реакций электрофильного замещеения и окисления. С другой стороны, наличие положительного мезомерного эффекта приводит к укорачиванию связи С-ОН, что приводит к снижению основных и нуклеофильных свойства фенольного гидроксила (по сравнению со спиртами) и делает практически невозможными реакции нуклеофильного замещения. Влияние заместителей на кислотность фенола. На кислотность фенола оказывают влияние заместители в ароматическом ядре. 1. При наличии в пара-положении электроноакцепторных заместителей (-NO2, -С1 др.) кислотные свойства усиливаются. 2. Если в п-положение ввести электронодонорные заместители (-NН2, -ОСН3 и др.), то происходит снижение кислотных свойств, поскольку уменьшается поляризация О-Н-связи, что затрудняет отрыв протона: 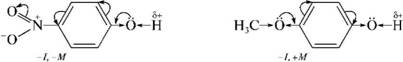 Рис. 24.12. Влияние заместителей в бензольном кольце на кислотные свойства фенола Получение простых и сложных эфиров. Фенолы, являясь слабыми основаниями, обладают и низкой нуклеофильностью. Реакции с участием фенолов в роли нуклеофила протекают с большим трудом. Значительно облегчает протекание реакции использование щелочной среды, в которой фенолы превращают в феноксид-ионы ArO- (феноляты), обладающие высокой нуклеофильностью. Получение фенолятов. Кислотность фенолов значительно выше, чем спиртов, поскольку феноксид-ионы в результате мезомерной делокализации отрицательного заряда стабильнее алкоксид-ионов: 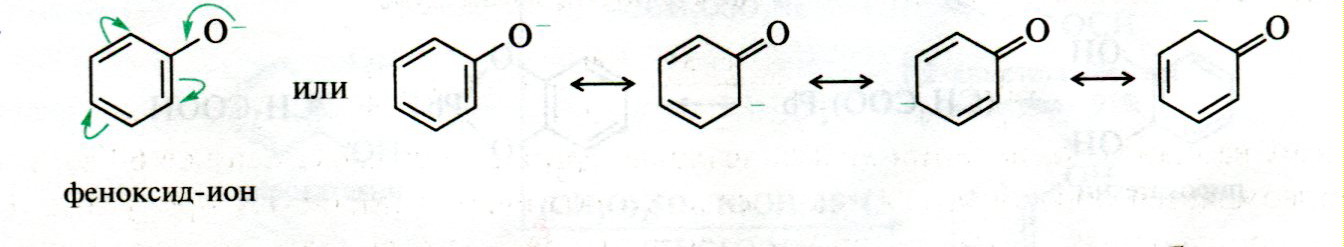 Рис. 24.13. Схема мезомерной делокализации заряда в феноксид-ионе Повышенная кислотность фенолов проявляется в их способности взаимодействовать с растворами щелочей: 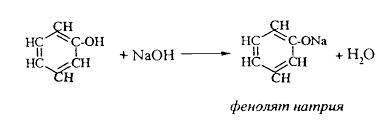 24.14. Схема реакции взаимодействия фенолов с растворами щелочей О-алкилирование (получение простых эфиров). Фенолы в щелочной среде под действием алкилирующих агентов (галогеналканов, диалкилсульфатов) превращаются в алкилариловые эфиры: 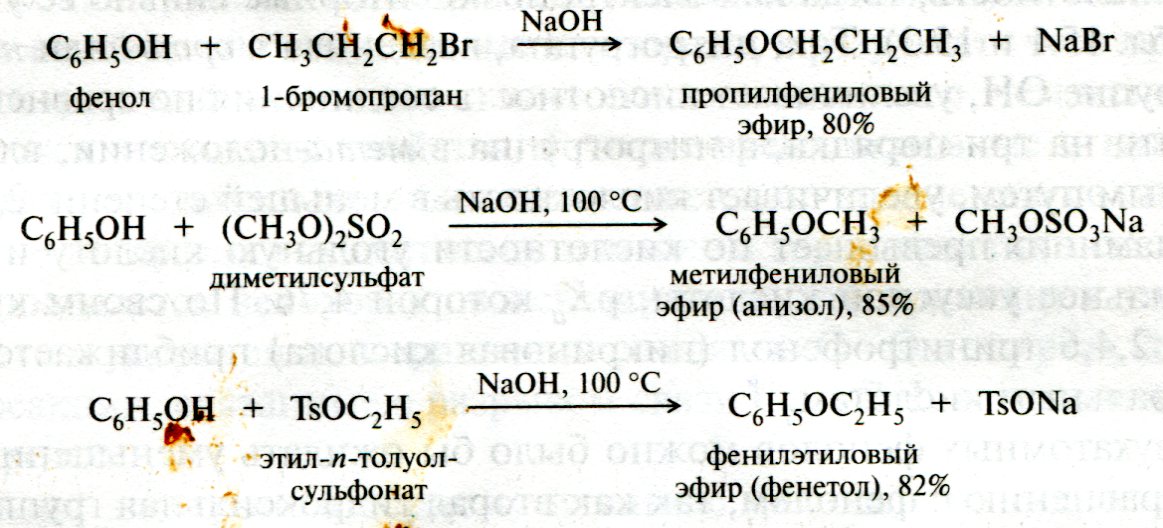 Рис. 24.15. Схема реакции О-алкилирования фенолов О-ацилирование (получение сложных эфиров). Гидроксильная группа фенолов ацилируется под действием активных ацилирующих агентов – хлорангидридов и ангидридов карбоновых кислот, в результате чего образуются сложные эфиры В связи с низкой нуклеофильностью фенолов реакции катализируют либо минеральными кислотами (серная, фосфорная) с целью активации ангидрида, либо основаниями с целью активации фенола (ацетат натрия, третичные амины): 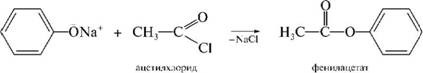 Рис. 24.16. Схема реакции О-ацилирования фенолов хлорангидридом 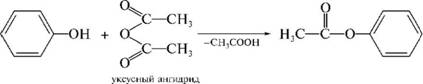 Рис. 24.17. Схема реакции О-ацилирования фенолов ангидридами карбоновых кислот !!! В отличие от спиртов фенолы не этерифицируются непосредственно карбоновыми кислотами. Реакции электрофильного замещения в ароматическом ядре. Фенольная гидроксильная группа является одним из сильнейших электронодонороных ориентантов I рода. Наличие гидроксильной группы в феноле значительно облегчает протекание типичных реакций электрофильного замещения по сравнению с бензолом. Галогенирование. Галогенирование фенолов протекает исключительно легко при комнатной температуре без катализаторов при действии бромной или хлорной воды: 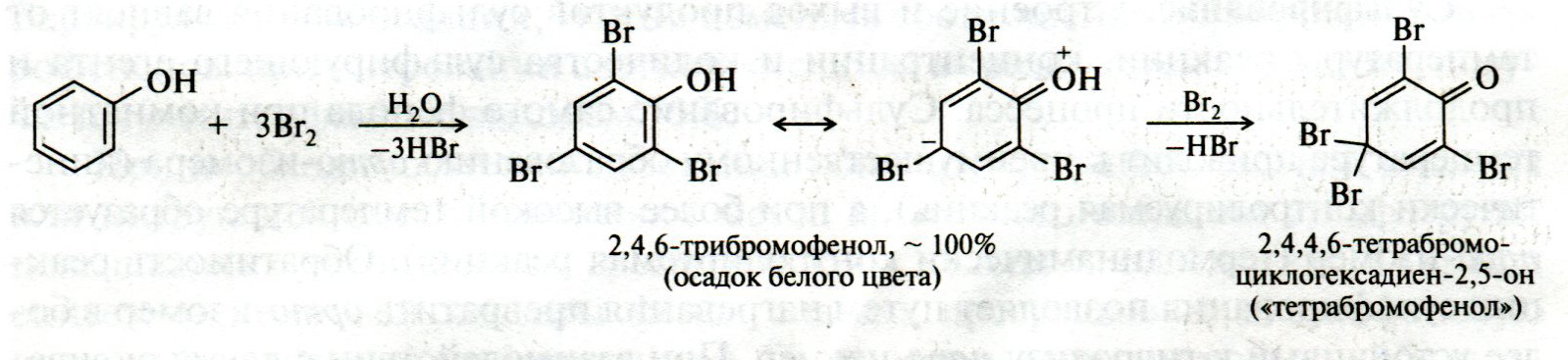 Рис. 24.18. Схема реакции галогенирования фенолов 1. Полярный растворитель (вода) сольватирует молекулу фенола, в результате чего сольватированная ОН-группа проявляет больший электронодонорный эффект, чем несольватированная. 2. Следствием этого является легкое, трудно контролируемое замещение атомов водорода сразу в трех положениях. 3. При избытке бромной воды реакция протекает дальше с образованием «тетрабромфенола» (осадок желтого цвета). 4. Эта реакция используется для качественного и количественного определения фенола. Чувствительность реакции такова, что позволяет обнаруживать фенол в концентрации 10-4 моль/л. Сульфирование. Строение и выход продуктов сульфирования зависят от температуры реакции, концентрации и количества сульфирующего агента. Сульфирование самого фенола при комнатной температуре приводит к преимущественному образованию орто-изомера, а при более высокой температуре образуется пара-изомер: 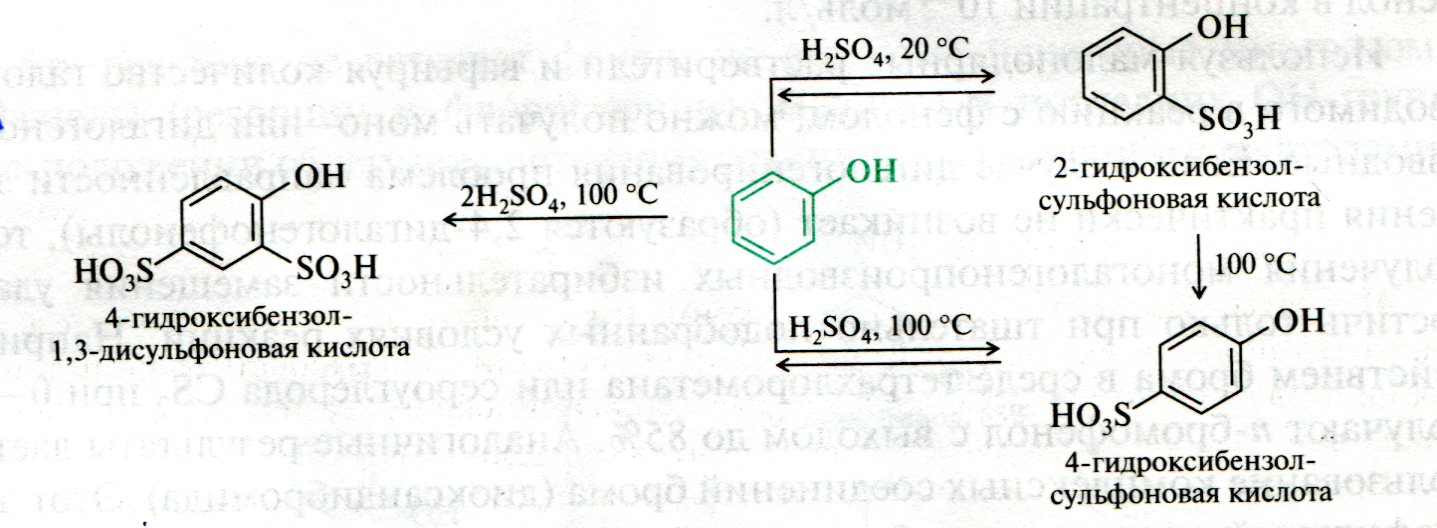 Рис. 24.19. Схема реакции сульфирования фенолов Обратимость реакции сульфирования позволяет путем нагревания превратить орто-изомер в более устойчивый к гидролизу пара-изомер. Нитрование. Фенолы очень легко вступают в реакцию нитрования. Так, при действии 20% азотной кислоты на фенол образуется смесь орто- и пара-нитрофенолов в соотношении 3 : 1. орто-Изомер, как более летучий из-за внутримолекулярной водородной связи, легко отделяется перегонкой с водяным паром: 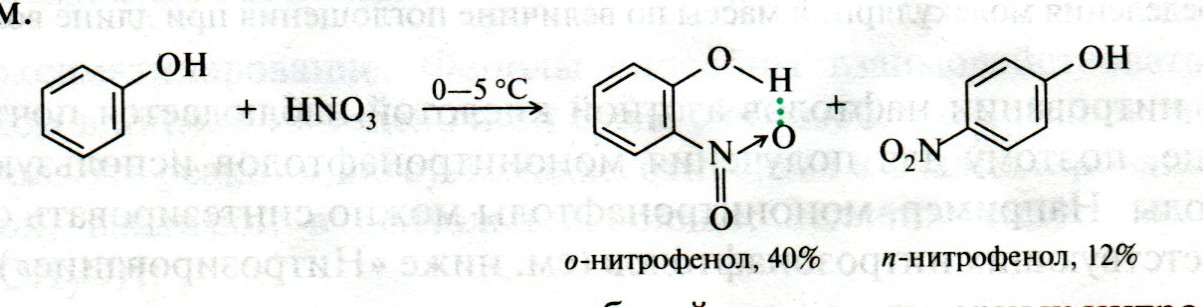 Рис. 24.20. Схема реакции нитрования фенолов Не смотря на легкость замещения, общий выход изомерных фенолов не высокий. Это объясняется склонностью фенолов к окислению даже при действии разбавленной азотной кислоты, что приводит к образованию смолоподобных продуктов окисления и полимеризации. С-алкилирование. Алкилирование фенолов в условиях реакции Фиделя-Крафтса затруднено тем, что кислоты Льюиса образуют с неподеленной парой электронов кислорода нереакционноспособный донорно-акцепторный комплекс. 1. Однако фенолы могут быть алкилированы спиртами и алкенами в присутствии серной или фосфорной кислоты. 2. Как и арены, они при этом легко подвергаются полиалкилированию, что приводит к образованию смеси моно-, ди- и триалкилфенолов. 3. Избежать полиалкилирования можно введением электроноакцепторной сульфогруппы, которая будет потом удалена из продукта алкилирования. Такой подход реализуется в способе получения антисептика тимола из м-крезола: 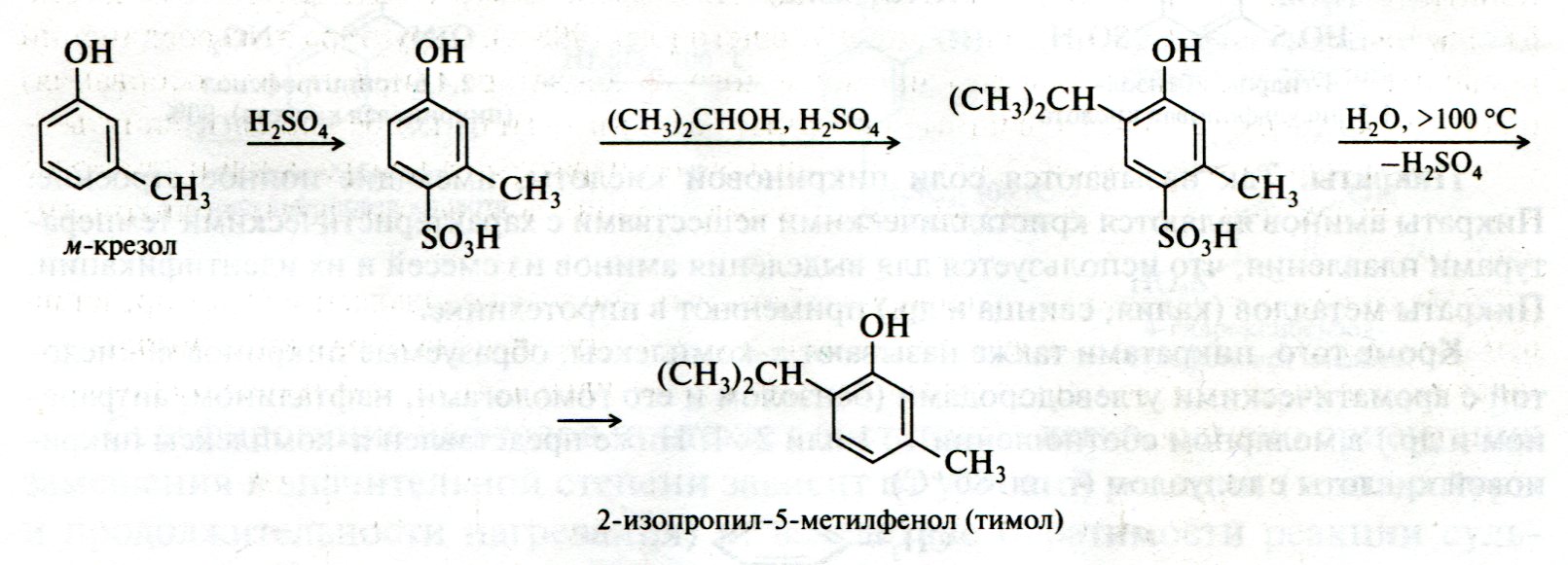 Рис. 24.21. Схема реакции С-ацилирования фенолов Конденсация фенолов с формальдегидом (гидроксимитилирование). Фенолы способны взаимодействовать с формальдегидом в кислой или щелочной среде с образованием гидроксибензиловых спиртов. Эта реакция тоже относится к С-алкилированию, только группой, вводимой в бензольное кольцо, является гидроксиметильная группа: 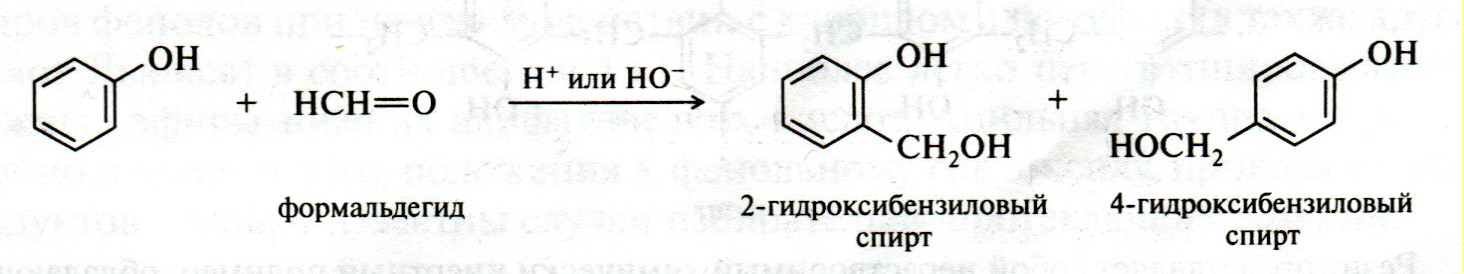 Рис. 24.22. Схема реакции гидроксиметилирования фенола Фенолформальдегидные смолы — синтетические смолы со свойствами реактопластов или термореактопластов. Формальдегидные смолы являются жидкими или твердыми олигомерными продуктами поликонденсации фенола с формальдегидом в щелочной или кислой среде (бакелиты, новолачные и резольные смолы), что соответственно влияет на их свойства. Механизм образования фенолформальдегидных смол весьма сложен и представлен схематически. В кислой среде при небольшом избытке фенола образуются новолаки – низкомолекулярные смолы, не содержащие гидроксиметильных групп в бензольном кольце: 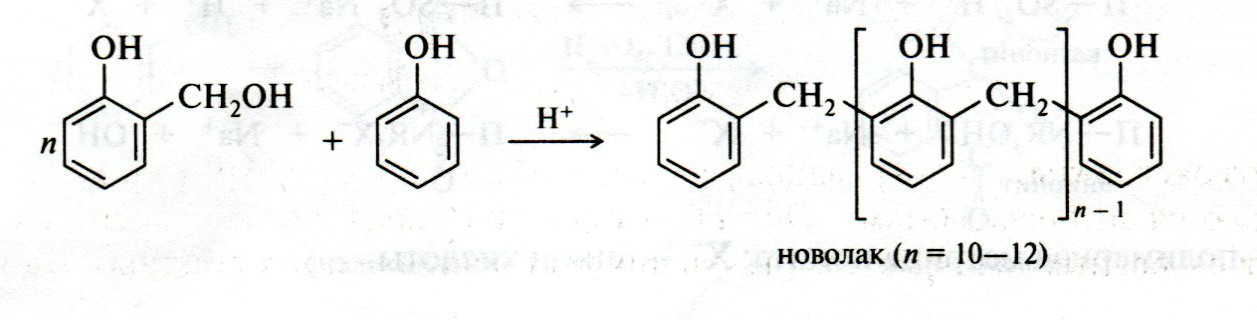 Рис. 24.22. Схема реакции полимеризации (поликонденсации) 2-метилгидроксифенола и фенола (синтез новолака) При взаимодействии фенола и формальдегида, взятых в количествах, близких к эквивалентным, или при избытке формальдегида образуется продукт поликонденсации – резол (1 стадия): 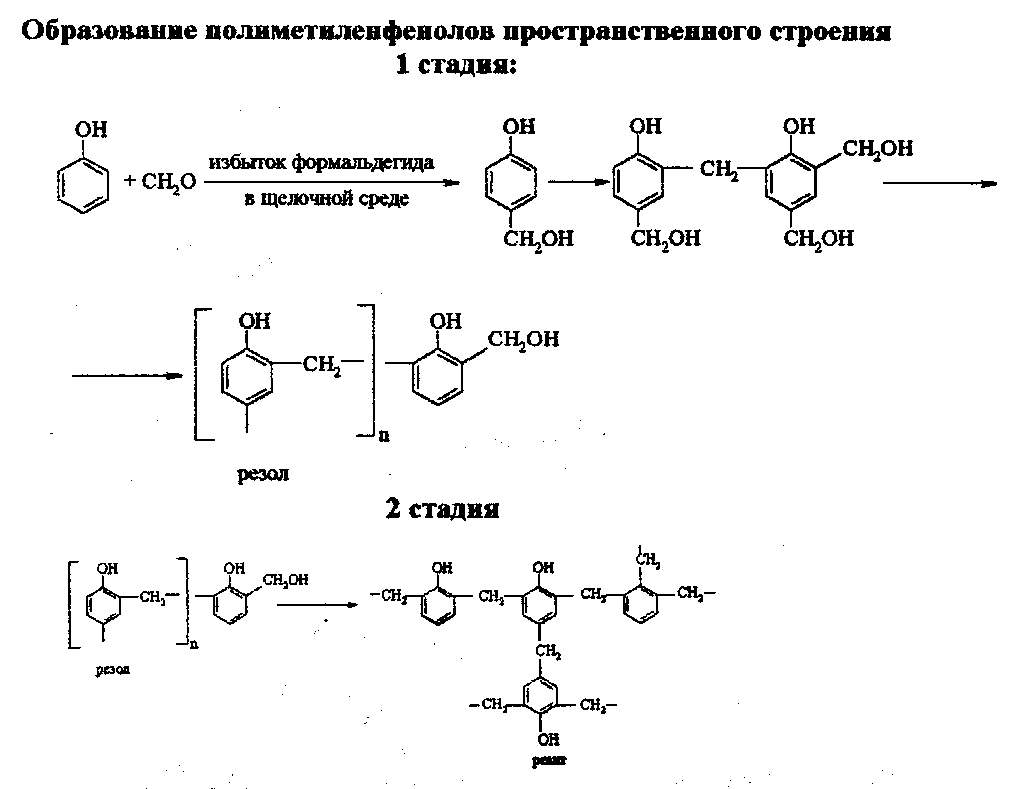 Рис. 24.22. Схема реакции синтеза резола (1-я стадия) и резита (2-я стадия) Химической особенностью резолов является возможность дальнейшей поликонденсации за счет алкилирования гидроксиметильными группами бензольных колец другой полимерной цепи. Такое сшивание цепей метиленовыми группами при 130º С-150ºС приводит к образованию сетчатой структуры фенолформальдегидной смолы, называемой резитом (2-я стадия). Фенолформальдегидные смолы относятся к дешевым полимерам и применяются для изготовления различных деталей во многих областях техники. Специфические реакции электрофильного замещения, характерные для фенолятов: получение карбоновых кислот (реакция Кольбе-Шмитта); альдегидов бензольного ряда (реакция Раймера-Тимона), азосочетание. Получение карбоновых кислот – карбоксилирование (реакция Кольбе-Шмитта). При действии диоксида углерода на твердые феноксиды щелочных металлов образуются фенолокислоты: 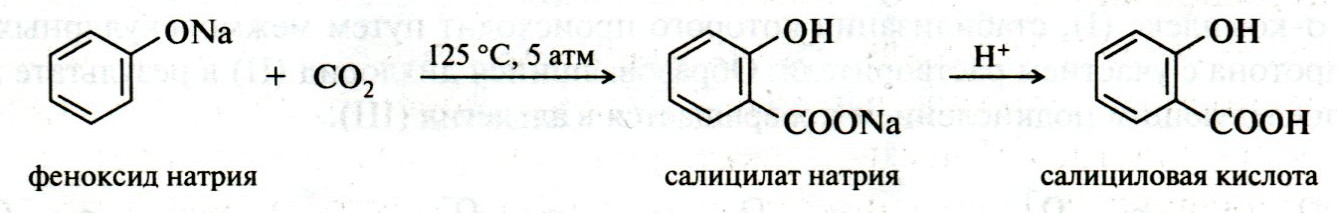 Рис. 24.23. Схема реакции карбоксилирования феноксидов щелочных металлов Эта реакция является основным способом получения салициловой (2-гидроксибензойной) кислоты. Реакция протекает по механизму электрофильного замещения. Слабый электрофил – диоксид углерода, являющегося нуклеофильной кислотой, взаимодействует с мягким основанным центром амбидентного феноксид-иона, находящегося в орто-положении. В результате образуется σ-комплекс, стабилизированный в виде хелата с участием иона натрия. Этот комплекс обычным путем (отщепление протона и возврат к ароматической системе) превращается в салицилат натрия: 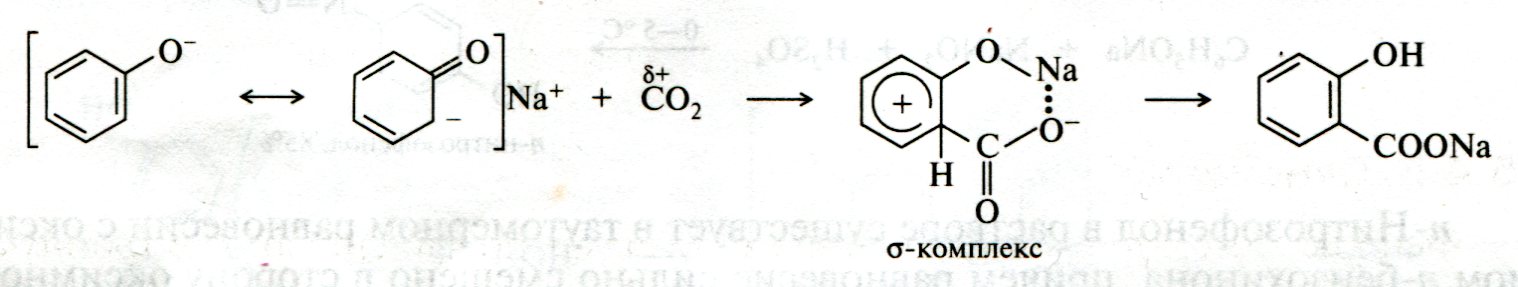 Рис. 24.23. Механизм реакции карбоксилирования феноксидов щелочных металлов Получение альдегидов бензольного ряда – формилирование (реакция Раймера –Тимона). При нагревании фенолов с хлороформом в водном или спиртовом растворе щелочи образуют ароматические гидроксиальдегиды. В этом превращении получаются орто-изомеры. Несмотря на невысокие выходы (около 50%), этот синтез применяется довольно широко, так как является прямым методом введения альдегидной группы в кольцо фенолов. Из самого фенола образуется салициловый альдегид: 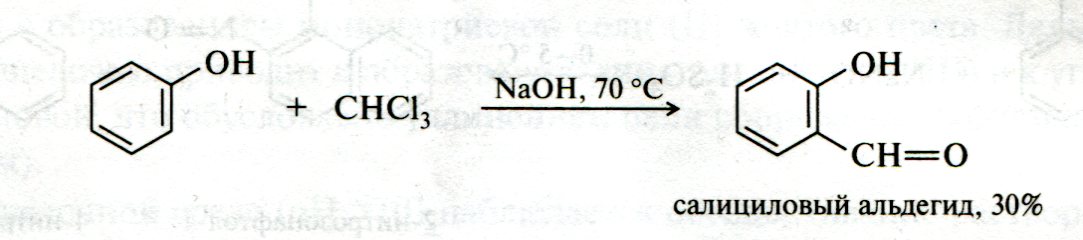 Рис. 24.24. Схема реакции формилирования фенолов Электрофильным агентом в этой реакции является синглетный дихлорокарбен :CCl2, образующийся при действии щелочи на хлороформ: 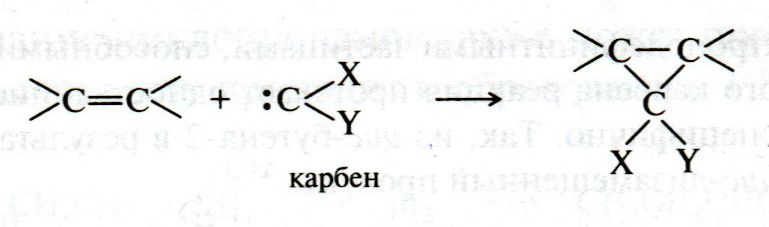 Рис. 24.25. Структурная формула синглетного дихлоркарбена Карбены высокореакционные частицы двухвалентного углерода общей формулы: :CXY (где X и Y – водород, галоген или органический заместитель), имеющий только 6 внешних электронов. Нейтральная неустойчивая молекула карбена существует в двух формах. В одной из них два внешних электрона спарены и находятся на одной орбитали, в другой – каждый электрон находится на собственной орбитали, и такой карбен представляет собой бирадикал. Первый карбен называется синглетным, второй - триплетным. К наиболее распространенным карбенам относится :СН2 (собственно карбен или метилен) и :СCl2 (дихлоркарбен). В результате электрофильной атаки феноксид-иона дихлоркарбеном возникает σ-комплекс (I), стабилизация которого происходит путем межмолекулярного переноса протона с участием растворителя. Образовавшийся дихлорид (II) в результате гидролиза с последующим подкислением превращается в альдегид (III): 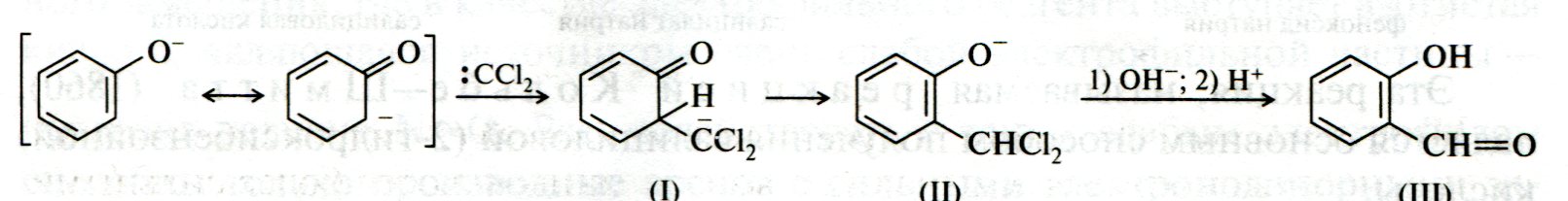 Рис. 24.25. Схема реакции формилирования фенолов Азосочетание. Взаимодействие диазосоединений с ароматическими аминами и фенолами, сопровождающееся образованием веществ, содержащих азогруппу —N=N—, связанную с двумя ароматическими радикалами, называют азосочетанием. Диазосоединения получили название диазосоставляющих реакции азосочетания, а амины или фенолы - азосоставляющей. Установлено, что реакция азосочетания протекает по механизму электрофильного замещения, в котором атакующим регентом выступает ион диазония (Аг—N=N), а субстратом - ароматические системы, содержащие в п- или о-положении заместители NH2, NHAlk, NHAr, N(Alk)2, NHSO3H, NHNO2, ОН и в некоторых случаях OAlk: 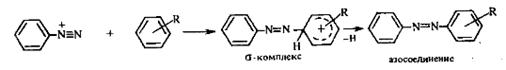 Рис. 24.25. Механизм реакции азосочетания Вследствие небольшой активности ионов диазония реакция протекает направленно с образованием п- или о-изомеров. Электроноакцепторные заместители в диазониевом ионе повышают его реакционную способность, а электронодонорные группы понижают ее. Окисление фенолов. Антиоксиданты. Фенолы, имеющие по сравнению с аренами повышенную электронную плотность окисляются довольно легко. Фенол уже при стоянии на воздухе приобретает розовую или красную окраску, обусловленную продуктами окисления. Окисление фенола сильными окислителями (триоксид хрома, хромовая смесь и др.) приводит к образованию, наряду с дру азосоставляющая гими продуктами, п-бензохинона: 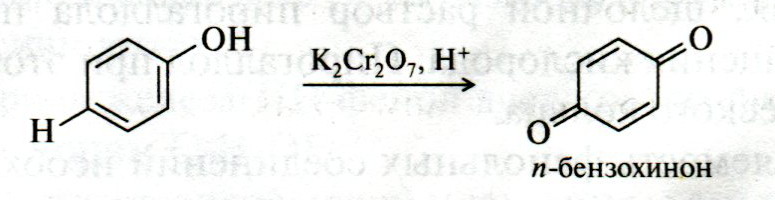 Рис. 24.26. Схема реакции окисления фенола до бензохинона Реакция окисления фенола протекает сложно, многоступенчато, и конечный результат определяется имеющимися в кольце заместителями и силой окислителя. Начальная стадия заключается в образовании феноксильного радикала, стабилизированного за счет резонансной изомерии: 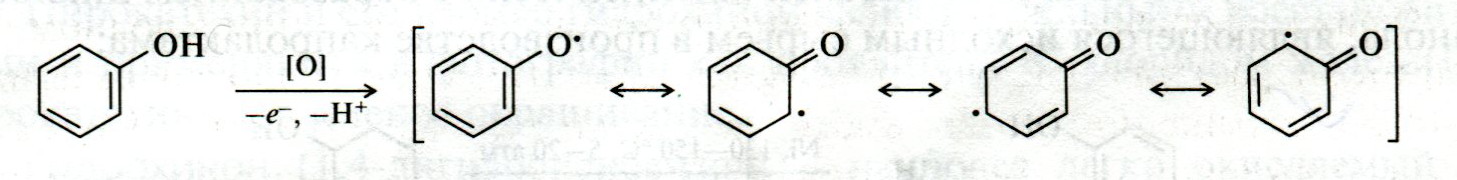 Рис. 24.27. Механизм реакции окисления фенола до бензохинона Феноксильный радикал затем подвергается дальнейшим превращениям и окисляется в бензохинон. Антиоксиданты. 2,4,6-Триалкилфенолы, особенно с третичными алкильными группами, являются оксиданнтами. Они предотвращают окислительные процессы путем связывания свободных радикалов, вызывающих рост цепи в цепных реакциях. Сами же оксиданты превращаются в стабильные и малоактивные свободные радикалы, которые существуют длительное время за счет распределения неспаренного электрона по всей ароматической системе и стерических препятствий рекомбинации: 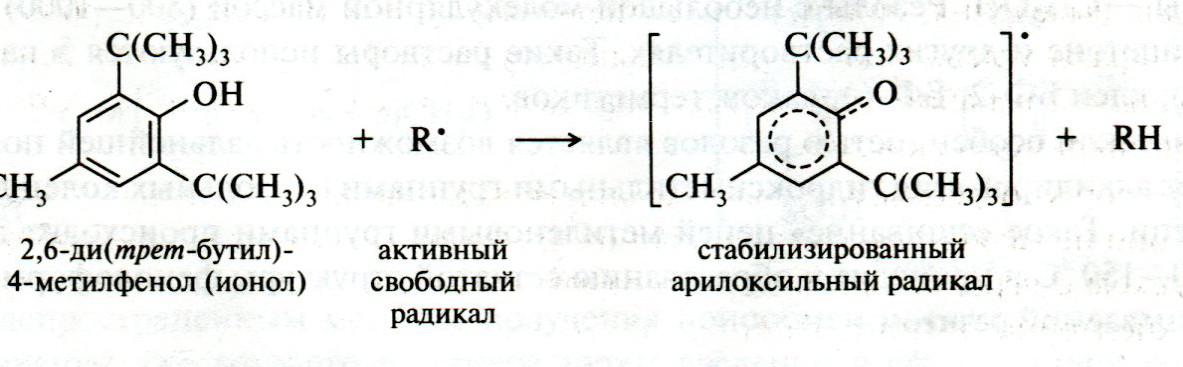 Рис. 24.27. Схема реакции связывания свободных радикалов триалкилфенолами (антиоксидантами) Восстановление. Фенол может быть восстановлен каталитически с образованием циклогексанола, являющегося исходным сырьем в производстве капролактама:  Рис. 24.28. Схема реакции восстановления фенола до циклогексанола |