|
|
Механизмы развития. Механизмы развития вторичного посттравматического иммунодефицита
Федеральное государственное бюджетноевоенное образовательное учреждениевысшего профессионального образования МО РФ Военно-медицинская академия имени С.М. Кирова
Кафедра патологической физиологии
Реферат на тему:
«Механизмы развития вторичного посттравматического иммунодефицита»
Выполнил:
студент 7 факультета, 3 курса, гр. 1837 б
Исмаилов Эльнур Арифович
Проверил:
Преподаватель Лавинская Н.Н
Санкт-Петербург
2021
Содержание
ВВЕДЕНИЕ 3
1 Вторичный посттравматический иммунодефицит 4
Общие сведения 4
Влияние цитокинов на иммунный статус 5
Влияние адренокортикотропного гормона на иммунный статус 9
Влияние катехоламинов на иммунный статус 12
Другие факторы, влияющие на развитие вторичного
посттравматического иммунодефицита 15
ЗАКЛЮЧЕНИЕ 16
СПИСОК ЛИТЕРАТУРНЫХ ИСТОЧНИКОВ 17
ВВЕДЕНИЕ
Иммунные дисфункции сопровождают различные патологические состояния. В условиях несостоятельности противоинфекционного иммунитета развиваются различные инфекционные заболевания и гнойно-септическая патология как особая форма ответа организма на патоген. При травмах возникает вторичный посттравматический иммунодефицит, усугубляющийся в процессе развития травматической болезни. В развитии данного иммунодефицита играет роль синтеза цитокинов, адренокортикотропного гормона, катехоламинов. Тема является актуальной, так как встречаемость данной иммунной дисфункции даже в мирное время достаточно высока (тяжелые обширные травмы, ожоги, объемные и длительные оперативные вмешательства, кровопотери и т.п.)
1 Вторичный посттравматический иммунодефицит
Общие сведения
Вторичные иммунодефициты – нарушения иммунной защиты организма, развивающиеся в постнатальном периоде в результате воздействия на организм различных внешних или внутренних факторов, механизмы которых напрямую не связаны с первичным поражением генетического аппарата. Например, при тяжелой черепно-мозговой травме (ТЧМТ) развивается вторичный посттравматический иммунодефицит [1,2].
Иммунологическую недостаточность при ТЧМТ нельзя рассматривать изолированно от системной воспалительной реакции, неизбежно развивающейся в ответ на любое травматическое воздействие. Нейроэндокринная взаимосвязь между центральной нервной системой и иммунной системой осуществляется с помощью многочисленных гормональных факторов и медиаторов, которые одновременно принимают активное участие в развитии системной воспалительной реакции. Нейровоспалительный ответ при ТЧМТ включает стереотипные реакции врожденной иммунной системы, направленные на ограничение распространения повреждения ткани и восстановление гомеостатического баланса организма. Иммунокомпетентные клетки, проникая в ткань мозга, принимают активное участие в нейровоспалении. Пусковым моментом местной воспалительной реакции является поступление внутриклеточных компонентов поврежденных клеток (DAMPs damage associated molecular patterns) в паренхиму мозга. Эти молекулы «опасности» способствуют активации клеток микроглии и астроцитов, которые начинают активно синтезировать провоспалительные цитокины и запускают продукцию целого каскада медиаторов, вызывая развитие воспалительной реакции на местном и системном уровнях [4].
В развитии вторичного посттравматического иммунодефицита важную роль играет нейрогуморальный фактор (синтез цитокинов, адренокортикотропного гормона, катехоламинов).
Влияние цитокинов на иммунный статус
Цитокины обладают аутокринным (влияние на саму клетку), паракринным (на рядом расположенные клетки) и эндокринным (дистантные или системные влияния) свойствами. Но в первую очередь они являются медиаторами межклеточного взаимодействия и способствуют привлечению периферических иммунных клеток в ткань мозга. В зависимости от степени участия в активации иммунокомпетентных клеток, цитокины принято подразделять на провоспалительные (IL1β, IL6, IL8, IL12, IL18, TNFα, IFNγ), противовоспалительные (IL4, IL10, TGF β) и регуляторные (IL2, IL4, IL5, IL7, IFNγ). Наиболее изучены эффекты провоспалительных цитокинов (TNFα, IL1β, IL6, IL8) [5]. Так, синтез интерлейкина-1β (IL1β) повышается в ткани мозга в течение первых трех часов после травматического воздействия, в связи с чем его относят к «ключевому» цитокину цереброваскулярной воспалительной реакции. Пик синтеза интерлейкина-6 (IL6) выявлен через 2-8 часов, фактор некроза опухоли-α (tumor necrosis factor alpha, TNF-α) – через 3-8 часов после повреждения ткани. Известно, что IL6 участвует в нейрогенезе, но он также необходим зрелым нейронам и глиальным клеткам в физиологичных условиях их функционирования. В привлечении иммунокомпетентных клеток в очаг повреждения важную роль отводят интерлейкину-8 (IL8), способствующему повышению проницаемости ГЭБ. При увеличении его синтеза накопление макрофагов и иммунокомпетентных клеток в поврежденной ткани мозга происходит быстрыми темпами [4, 5]. Хемокины активно экспрессируются всеми клетками центральной нервной системы, в том числе и нейронами головного мозга (CX3CL1, CXCL14/BRAK, CCL2059, CCL21, CXCL12/SDF-1 и CCL2/MCP-1) [5]. Таким образом, с помощью цитокинов и химокинов в мозге формируется сложная коммуникационная сеть между нейронами и клетками микроокружения, способствующая привлечению иммунокомпетентных клеток, основная задача которых состоит в развитии местного воспаления, целью которого является регенерация поврежденной ткани мозга. Одновременно включаются механизмы ограничения избыточности воспалительной реакции, в которых важная роль принадлежит антивоспалительным цитокинам, в том числе интерлейкину-10 (IL10).
В результате повреждения ГЭБ первичные и вторичные медиаторы воспаления неизбежно поступают в кровоток способствуя развитию системной воспалительной реакции. Доказательства преимущественного синтеза провоспалительных цитокинов в очаге повреждения при травматическом повреждении головного мозга приведены в ряде исследований, выявивших больший уровень цитокинов в спиномозговой жидкости в сравнении с их концентрацией в сыворотке крови [3,5]. В то же время установлено формирование дополнительного экстратекального пула цитокинов за счет их синтеза иммунокомпетентными клетками. Были выявлены перекрестные связи: интратекальное введение IL1β способствовало повышению IL6 в системном кровотоке [4]. Имеется экстратекальный синтез про- и противовоспалительных цитокинов при ТЧМТ. Попадая в систему циркуляции, цитокины одновременно запускают системную воспалительную реакцию и активацию неспецифического иммунного ответа: повышение белков острой фазы воспаления, увеличение уровня лейкоцитов и т.д. В настоящее время накопилось большое количество убедительных фактов, указывающих на неизбежность развития персистирующей воспалительной реакции при ТЧМТ. Адекватный и регулируемый воспалительный ответ необходим организму для защиты от инфекций в условиях нарушения целостности тканей и гематоэнцефалического барьера. В системном кровотоке при ТЧМТ выявлен гетерогенный набор белков DAMPs: митохондриальные белки (mtDAMPs), ДНК ткани головного мозга, нуклеарный протеин HMGB1 (high mobility group box-1), белок S100. Поступление молекул DAMPs в кровоток является ключевым связующим звеном между повреждением, системным воспалительным ответом и синдромом полиорганной недостаточности.
Множественные провоспалительные сигналы DAMPs путем связывания толл_подобных рецепторов клеток TLR4 и TLR2 (Toll-like receptor) обеспечивают активацию врожденного иммунного ответа. Эволюционно рецепторы клеток TLR4 возникли для связывания липополисахарида клеточной стенки грамотрицательных бактерий, а TLR2 – для соединения с молекулярными структурами грамположительных бактерий, включая пептидогликаны, липотейхоевую кислоту, а также зимозана клеточной стенки дрожжей. Но одновременно эти рецепторы принимают участие в связывании DAMPs и запускают сложный транскрипционный процесс ранних провоспалительных генов цитокинов. Кроме того, молекулы DAMPs выступают мощными активаторами нейтрофилов, макрофагов и лимфоцитов, вызывают дегрануляцию нейтрофилов и выброс активных форм кислорода, что объясняет наличие оксидативного стресса при ТЧМТ. Особенностью иммуносупрессии при ТЧМТ является снижение преимущественно Т-лимфоцитов, маркеров активации лимфоидных клеток и синтеза IgG [4]. Развитие иммунологической недостаточности при травматическом повреждении головного мозга в первую очередь связано с изменением центральной регуляции иммунокомпетентных органов. Так, в течение первых суток после травмы была выявлена зависимость величины внутричерепного давления с уровнем провоспалительных цитокинов и снижением числа лимфоидных клеток в кровотоке. При этом характерными особенностями иммуносупрессии при ТЧМТ являются снижение преимущественно Т-субпопуляций лимфоцитов, переключение иммунного ответа в направлении Th1>Th2. Объяснение этого феномена основывается на концепции баланса Th1/Th2 клеток, предложенной Mos-mann T.R. и соавторами в 1986 году. В соответствии с этой теорией, тип иммунного ответа определяется активацией клонов Th1- и Th2-лимфоцитов (рис. 1), различающихся по синтезу регуляторных цитокинов, обеспечивающих направление иммунного ответа по клеточному или гуморальному типу. Активация Th1-лимфоцитов, секретирующих IL2 и IFNγ, способствует развитию клеточного иммунного ответа, тогда как синтез Th2-цитокинов стимулирует гуморальное звено иммунитета. При этом Тh1- и Тh2-цитокины обладают взаимоингибирующим влиянием. Интерлейкин-12 является чрезвычайно сильным индуктором синтеза IFNγ и ингибирует синтез IL4, важного для дифференцировки и созревания Th2-клеток. В нормально функционирующем организме имеется баланс во взаимодействии между Тh1- и Тh2-цитокинами, но при ТЧМТ он смещается в направлении Th1>Th2. Изменение синтеза регуляторных цитокинов приводит к неблагоприятным последствиям функционирования иммунной системы в целом. При ТЧМТ в первые сутки у умерших пациентов отмечалось увеличение уровня IL12 в сыворотке крови [4], в то время как у выживших пациентов, напротив, отмечалось снижение IL12 наряду с IFNγ.
Снижение регуляторных цитокинов при ТЧМТ ограничивает способность Th-лимфоцитов к дифференцировке, а снижение уровня IL2 включает программы апоптоза зрелых Т-клеток. Показано, что недостаточность Т- и В-клеточного звеньев иммунитета опосредована повышением провоспалительных и недостаточностью Th1- и Th2-регуляторных цитокинов. Поскольку в защите организма от инфекций ведущая роль принадлежит Th1-хелперам и моноцитам/макрофагам, то переключение пролиферации лимфоцитов в направлении Th1>Th2 не является оптимальным при ТЧМТ, так как ведет к понижению функциональной активности фагоцитарных клеток. Поскольку последние играют ведущую роль в элиминации условно-патогенных бактерий, изменение условий их функционирования является причиной развития гнойно-воспалительных осложнений при критических состояниях.
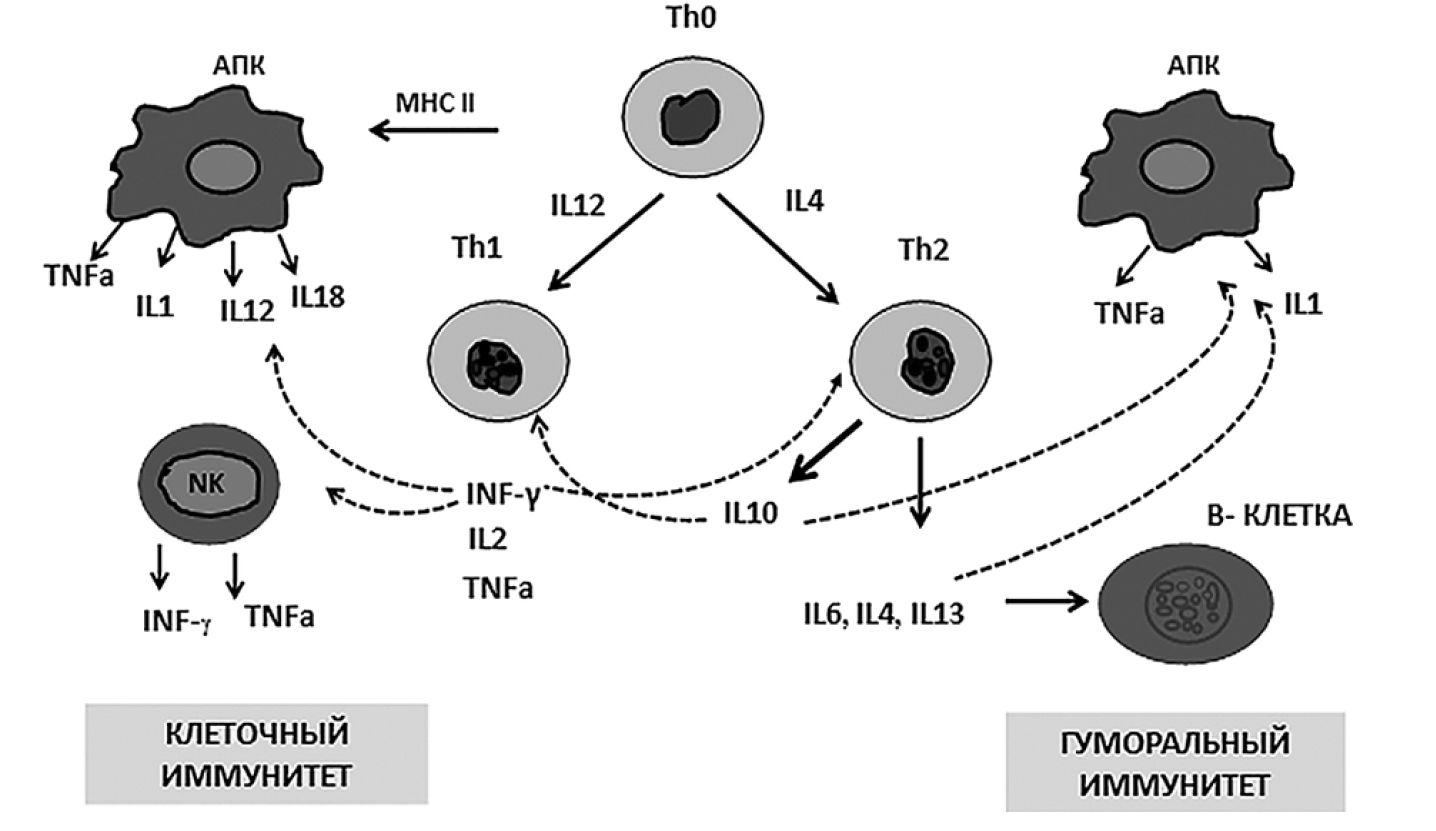
Рис. 1. Роль цитокинов в дифференцировке лимфоцитов при развитии Th1 и Th2 иммунного ответа [4].
Влияние адренокортикотропного гормона на иммунный статус
Немаловажную роль в регуляции числа лимфоидных клеток при ТЧМТ отводят гормональному влиянию. Высокие уровни кортизола и глюкокортикоидных гормонов также приводят к развитию иммуносупрессии.
Глюкокортикоиды снижают активность дендритных клеток путём:
ингибирования их созревания;
посредством нисходящей регуляции экспрессии молекул МНС II, липид-представляющих молекул CD1a, ко-стимулирующих молекул (например, CD80 и CD86) и провоспалительных цитокинов (таких, как IL-12 и TNF);
стимуляции экспрессии противовоспалительных цитокинов, таких как IL-10
Глюкокортикоиды также могут тормозить активацию Т-лимфоцитов, вмешиваясь в Т-клеточный сигналинг. Механизмы, вовлечённые в глюкокортикоид-опосредованное снижение TCR-сигналинга, включают нисходящую регуляцию FOS и нарушение активности AP-1, NF-kB и NFAT [6]
Хотя суммарный эффект глюкокортикоидов уменьшает активность Т-лимфоцитов, некоторые исследования говорят о том, что глюкокортикоиды преимущественно подавляют реакцию Th1- и Th17-лимфоцитов, при этом сохраняя, а иногда даже стимулируя функцию Th2-лимфоцитов и регуляторных Т-клеток (Treg). Механизмы предполагаемой глюкокортикоид-индуцированной Th2-поляризации включают down-регуляцию антигенпрезентирующими клетками продукции цитокина IL-12, стимулирующего Th1-клетки, снижение экспрессии рецептора к IL-12 на мембране Т-лимфоцитов, ингибирование мастер-фактора транскрипции Th1-лимфоцитов T-bet (TH1 cell master transcription factor Tbet) и повышение продукции Th2-лимфоцитами классических цитокинов Th2-типа (IL-4, IL-10 и IL-13)
Влияние на развитие и выживание В-лимфоцитов. В некотором роде действие глюкокортикоидов на тимопоэз отражается и на В-лимфопоэзе. Юные В-лимфоциты более чувствительны ко глюкокортикоид-индуцированному апоптозу, чем зрелые В-лимфоциты [8], и адреналэктомия или введение антагониста рецепторов глюкокортикоидов мифепристона приводят к росту популяции юных В-лимфоцитов в костном мозге. В-лимфоциты экспрессируют рецепторы глюкокортикоидов в процессе созревания, и эти рецепторы оказывают влияние на несколько факторов транскрипции, исходящих от сигнальных путей рецепторов В-лимфоцитов (например, AP1, NFκB и NFAT), что повышает возможности глюкокортикоидов оказывать влияние на селекцию В-лимфоцитов.
На наш взгляд, механизмы взаимодействия между сигнальными путями рецепторов глюкокортикоидов и рецепторов В-лимфоцитов (BCR) пока недостаточно изучены. Намёки на прямое влияние глюкокортикоидов на функции В-лимфоцитов можно увидеть в исследованиях мышей с отсутствием гена-мишени глюкокортикоидов Gilz. В ответ на стимуляцию BCR клетки с отсутствующим Gilz демонстрируют повышенный пролиферативный ответ, и с возрастом у Gilz-нокаутных мышей развивается волчаночноподобный синдром. Мыши с отсутствием Gilz в В-лимфоцитах не демонстрируют аутоиммунный фенотип глобального (т.е. во всех клетках, прим. перев.) дефицита Gilz, что показывает выходящую за пределы В-лимфоцитов роль GILZ в продукции аутоантител; однако, мыши с избирательным дефицитом экспрессии GILZ в В-лимфоцитах демонстрируют системное увеличение числа В-лимфоцитов, что подтверждает специфичную роль сигналинга рецепторов глюкокортикоидов в выживании и/или гомеостазе В-лимфоцитов.
Влияние на продукцию антител. Пока не удалось достичь общего согласия насчёт влияния глюкокортикоидов на гуморальный иммунитет, хотя в общем фармакологическое лечение использованием глюкокортикоидов связано со снижением концентрации иммуноглобулинов в крови. Лишь в нескольких исследованиях были рассмотрены последствия долгосрочного лечения глюкокортикоидами при вакцинации людей и было сообщено о небольшом воздействии (если таковое имелось) на титры антител. Тем не менее некоторое внимание получило влияние глюкокортикоидов на продукцию IgE, и некоторые клинические и in vitro исследования подтверждают, что глюкокортикоиды могут при определённых условиях способствовать продукции этого изотипа иммуноглобулинов. В исследовании пациентов с астмой, получавших терапию глюкокортикоидами, концентрации IgE в сыворотке возрастали, в то время как концентрации других иммуноглобулинов не изменялись или уменьшались [5,6]. Предлагаемые механизмы глюкокортикоид-опосредованного повышения продукции IgE включают прямое влияние на B-клетки путём переключения класса изотипов совместно с ИЛ-4, а также посредством непрямых эффектов на В-лимфоциты через действие глюкокортикоидов на Т-лимфоциты и моноциты.
Характерной особенностью глюкокортикоидов является иммунодепрессивная активность. Иммунодепрессивные свойства глюкокортикоидов связаны с подавлением разных этапов иммунной реакции: торможения миграции стволовых клеток костного мозга и В-лимфоцитов, подавления активности Т- и B-лимфоцитов, а также угнетения высвобождения цитокинов (ИЛ-1, ИЛ-2, интерферона-гамма) из лейкоцитов и макрофагов. Кроме того, глюкокортикоиды снижают образование и увеличивают распад компонентов системы комплемента, блокируют Fc-рецепторы иммуноглобулинов, подавляют функции лейкоцитов и макрофагов.
Влияние катехоламинов на иммунный статус
Катехоламины оказывают прямое влияние не только на дифференцировку и пролиферацию лимфоцитов, но также на синтез цитокинов и выраженность миграции иммунокомпетентных клеток. Были выявлены различия в степени иммуносупрессирующего потенциала для Т-и В-лимфоцитов, связанные с неравномерностью симпатической иннервации лимфатических органов: зоны созревания T-клеток хорошо иннервированы, а фолликулярные области развития B-лимфоцитов иннервированы в меньшей степени [7,8]. Открытие воспринимающих рецепторов на мембране лимфоцитов объяснило суть механизма регулирования их функциональной активности катехоламинами.
Норадреналин и адреналин проявляют свои эффекты на лимфоидные клетки через адренергические рецепторы α и β (α-АР и β-АР). Установлено, что β-АР имеют три подтипа (β1, β2, β3), тогда как α-АР – два подтипа (α1, α2). Цитотоксический эффект катехоламинов на лимфоциты обусловлен их взаимодействием с β-АР [4], оказывая влияние на динамику созревания лимфоидных клеток, вплоть до включения процессов апоптоза через NF-κB-зависимые механизмы. Плотность расположения β-АР на лимфоцитах располагается в следующем порядке: NK-клетки > моноциты/макрофаги > T-супрессоры B-клетки > Th-клетки. В экспериментальном исследовании Sanders V.M. и соавт. показана экспрессия β2-АР на Th1-клетках, но не на Th2-лимфоцитах. Вышеперечисленные феномены объясняют модуляцию лимфоцитов в направлении Th1→Th2 в ответ на повышение уровня катехоламинов при ТЧМТ. Кроме того, блокирование катехоламинами экспрессии IL12 является еще одним фактором направления дифференцировки Th1→Th2 иммунного ответа [2]. По мнению Elenkov I.J. и соавт., ведущим механизмом блокирования синтеза IL12 и IL10 при ТЧМТ является стресс-индуцированный выброс глюкокортикоидов и катехоламинов. От уровня катехоламинов зависит также скорость миграции лимфоидных клеток в очаг повреждения, запуск программы апоптоза лимфоцитов, экспрессия генов раннего реагирования на стресс, сопровождающаяся синтезом белков острой фазы воспаления и цитокинов. Характерными особенностями иммуносупресии при ТЧМТ являются: большее снижение экспрессии молекул дифференцировки на CD4+-клетках (Т-хелперы) в сравнении с CD8+-лимфоцитами (Т-супрессоры), изменение экспрессии главного комплекса гистосовместимости I и II типа на мембранах клеток, большая чувствительность к процессам апоптоза Т-хелперов. Наряду с количественным изменением Т-клеток, были выявлены и их функциональные дефекты. Quattrocchi и соавт. Продемонстрировали снижение цитотоксической активности лимфокин-активированных киллеров (ЛАК-клеток) при ТЧМТ. Самыми «чувствительными» клетками иммунной системы к экстремальным воздействиям признаны NK-клетки, что объясняется наибольшим числом β2-АР на их поверхности. Поскольку на ранних стадиях воспаления NK-лимфоциты обладают способностью лизировать измененные клетки при отсутствии молекул гистосовместимости HLA-I (Human leukocyte antigens-I) и независимо от наличия антител и комплемента, то их снижение обуславливает повышенную восприимчивость пациентов к инфекциям, связанным с оказанием медицинской помощи [4,6]. При ТЧМТ выявлено абсолютное и относительное снижение количества NK-лимфоцитов в кровотоке. Кроме того, активность NK-клеток регулируется синтезом IL12 и IFNγ, которые крайне важны для их жизнеспособности. Последние годы характеризуются накоплением новых данных о механизмах развития иммуносупрессии при ТЧМТ. На рисунке 2 представлена схема, объединяющая известные механизмы иммунологической недостаточности, наряду с новыми открытиями в этой области. В патогенезе иммуносупрессии при ТЧМТ отводится немаловажная роль супрессорным клеткам миелоидного происхождения (MDSC – myeloid_derived suppressor cells). Повышение уровня хемокинов и цитокинов (TNF-α, IFNγ, IL1β, IL6 и TGF-β) способствует ускорению экспансии MDSC в костном мозге и приводит к их накоплению на периферии. Эта гетерогенная популяция незрелых миелоидных клеток обладает супрессорной активностью в отношении Т-лимфоцитов, дендритных и натуральных киллерных клеток. Выброс глюкокортикоидов и катехоламинов стимулирует пролиферацию и накопление миелоидных супрессорных клеток в кровотоке, которые способны снижать функциональную способность Т-лимфоцитов путем уменьшения аргинина и синтеза активных форм кислорода при ТЧМТ.
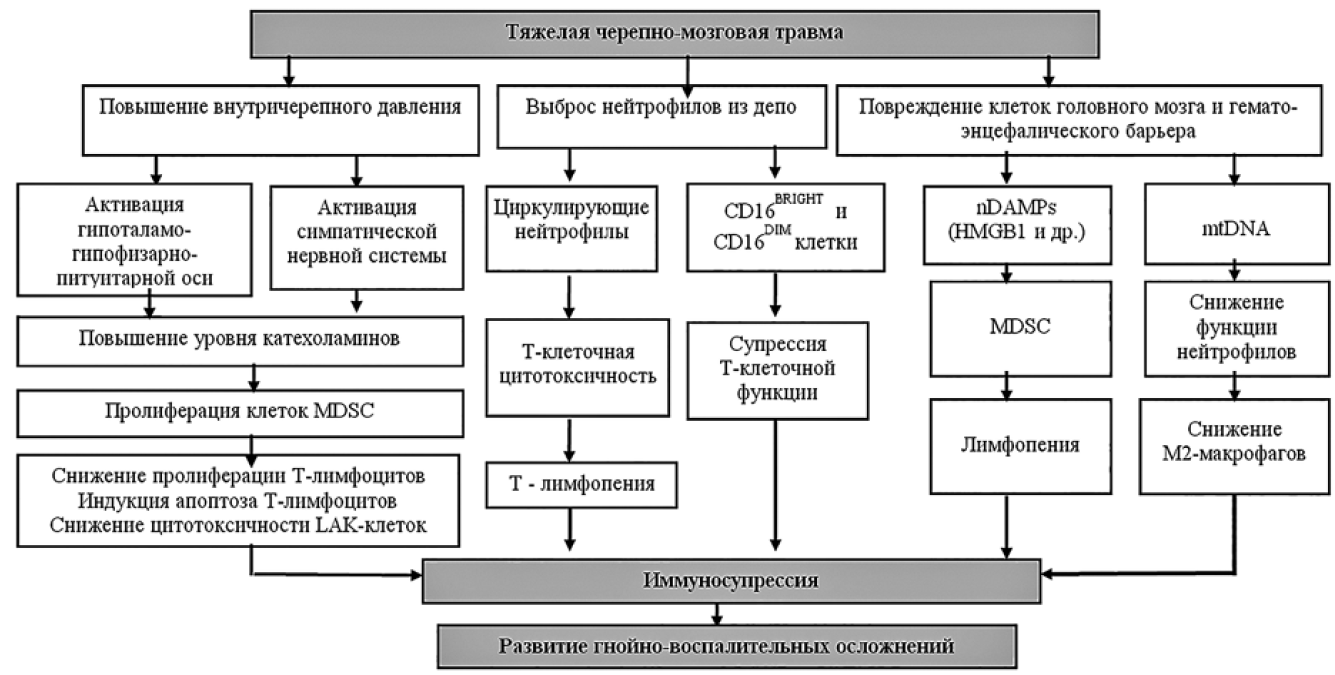
Рис. 2. Механизмы развития иммуносупрессии при тяжелой черепно-мозговой травме [4].
1.5 Другие факторы, влияющие на развитие вторичного посттравматического иммунодефицита
Дополнительными факторами, вносящими свой вклад в пролонгирование посттравматического иммунодефцита, являются: действие анестетиков, переливание крови и ее компонентов, недостаточность послеоперационного питания. Воспалительная реакция при ТЧМТ играет двойственную роль. С одной стороны, адекватный и контролируемый воспалительный ответ необходим для удаления клеточного детрита и запуска репаративных процессов в ткани мозга, с другой – персистирующая системная воспалительная реакция приводит к прогрессированию вторичных повреждений и развитию полиорганной недостаточности. Ограничение избыточности воспалительного ответа является важным патогенетическим условием для функционального восстановления организма. Поэтому в ответ на развитие системной воспалительной реакции активируются противовоспалительные механизмы, направленные на защиту клеток от влияния медиаторов воспаления, имеющих тканевой разрушительный потенциал [7]. Активация парасимпатической нервной системы при ТЧМТ вносит свой вклад в поддержание иммуносупрессии. Снижение способности моноцитов крови к антигенной презентации и уменьшение синтеза противовоспалительного цитокина IL10 способствуют поддерживанию воспалительного потенциала [8].
В целом быстрое развитие иммуносупресии, высокий уровень провоспалительных и низкий уровень антивоспалительных цитокинов способствуют формированию гнойно-воспалительных осложнений и неблагоприятному исходу [3,4,7]. С этих позиций, увеличение активности n. vagus в результате повышения внутричерепного давления при ТЧМТ несет в себе механизм отрицательной обратной связи, обеспечивая поддержание иммуносупрессии и одновременно ограничивая развитие системного воспалительного ответа.
ЗАКЛЮЧЕНИЕ
Механизмы развития иммуносупрессии многофакторны и включают стресс-индуцирующее изменение регуляции нейрогуморальной оси «центральная нервная система – иммунная система». Выброс катехоламинов, глюкокортикоидов и цитокинов способствует не только уменьшению циркулирующего пула лимфоцитов в кровотоке, но и изменению их функциональной активности. В целом снижение числа лимфоцитов при вторичном посттравматическом иммунодефиците обусловлено их миграцией в очаг повреждения, гибелью клеток путем апоптоза и замедлением их созревания в лимфопролиферативных органах. Развитие иммуносупрессии создает благоприятные условия для присоединения госпитальных инфекций и активации условно-патогенной микрофлоры в результате транслокации через поврежденную стенку кишечника.
СПИСОК ЛИТЕРАТУРНЫХ ИСТОЧНИКОВ
Патофизиология. Клиническая патофизиология: учебник / под редакцией В.Н. Цыгана // СПб.: СпецЛит — 2018. — 430 c.
Патофизиология / под редакцией В.Ю. Шанина // СПб.: ЭЛБИ-СПб — 2005. — 639 с.
Патофизиология. / П.Ф. Литвицкий // М.: ГЭОТАР-Медиа — 2016. — 624 с.
Борщикова Т.И., Епифанцева Н.Н., Кан С.Л. Механизмы формирования вторичной иммунологической недостаточности при тяжелой черепно-мозговой травме / Медицина в Кузбассе // 2018. — №3 — С. 58-65
Шанин С.Н., Фомичева Е.Е., Филатенкова Т.А. Коррекция нарушений нейроиммунных взаимодействий при экспериментальной черепно-мозговой травме препаратом рекомбинантного интерлейкина-2 / Медицинская иммунология // 2018. — №2 — С. 171-178
Мамонов И.А. Особенности состояния клеточного звена иммунитета до и после эндопротезирования крупных суставов / Саратовский научно-медицинский журнал // 2016. — №2 — С. 182-185
Жаркова И.В., Кабирова М.Ф., Герасимова Л.П. Локальный цитокиновый статус пациентов с хронической механической травмой слизистой оболочки рта, страдающих сахарным диабетом 1 типа / Проблемы стоматологии // 2018. — №1 — С. 16-20
Хатьков И.Е., Барсуков Ю.А., Атрощенко А.О. Иммунологические особенности малоинвазивных лапароскопических операций / Анналы хирургии // 2012. — №1 — С. 15-20
|
|
|
 Скачать 0.67 Mb.
Скачать 0.67 Mb.