Методичка - нефротический синдром, амилоидоз. Методические указания
 Скачать 154.5 Kb. Скачать 154.5 Kb.
|

|
МЕТОДИЧЕСКИЕ УКАЗАНИЯ Для самостоятельной подготовки к практическому занятию для студентов VI курса лечебного факультета Тема: Дифференциальная диагностика заболеваний, сопровождающихся нефротическим синдромом. Дифференциальная диагностика и дифференцированный подход к лечению различных типов амилоидоза. Цели практического занятия: - контроль исходного уровня знаний студентов по тест-вопросам; - разбор вопросов, которые остались неясными после самоподготовки к практическому занятию; - приобретение навыков и умений по сбору жалоб, анамнеза, объективному исследованию, дифференциальной диагностике у больных с нефротическим синдромом; - приобретение навыков и умений по проведению и интерпретации общеклинических, биохимических и инструментальных исследований; - контроль знаний и умений, навыков, приобретенных студентами на практическом занятии. Самоподготовка к практическому занятию Цель самоподготовки: после самостоятельного изучения материала студент должен знать: - причины и патогенез нефротического синдрома; - план диагностического поиска при нефротическом синдроме; - принципы этиопатогенетической и симптоматической терапии нефротического синдрома; - этиологию, патогенез, эпидемиологию амилоидоза; - биохимическую классификацию амилоидоза (по ВОЗ, 1993 г) и формулировку диагноза; - клинические варианты: идиопатический, вторичный, наследственный, гемодиализный; - значение пункционной биопсии почек, морфологического исследования слизистой десны и прямой кишки. Должен уметь: - проводить дифференциальную диагностику заболеваний, протекающих с нефротическим синдромом; - диагностировать различные клинические варианты амилоидоза, стадии вторичного амилоидоза; - назначать лечение больным амилоидозом в зависимости от клинического варианта и стадии заболевания. Рекомендуемая литература: Основная:
Дополнительная:
Вопросы для самоподготовки и самоконтроля
Письменное задание
Поэтапное выполнение работы на практическом занятии
Клинические задачи: Задача № 1. 43-летняя женщина отметила появление отеков на ногах, увеличение массы тела на 12 кг за 3 месяца. Прежде считала себя здоровой. Объективно: отеки нижних конечностей до средней трети бедра, рыхлые, кожные покровы бледные. В легких дыхание жесткое. Тоны сердца приглушены. ЧСС 90 в мин. АД 110/70 мм рт. ст. лежа, 90/70 мм рт. ст. стоя. Живот мягкий, безболезненный. Печень и селезенка не увеличены. Общий анализ крови: Hb 125 г/л, Le 7,7х109/л, СОЭ 30 мм/ч. Общий анализ мочи: уд. вес 1022, белок 32 г/л, Le 3-4 в п/зр., Er 1-2 в п/зр. В анализах крови холестерин 8,5 ммоль/л, общий белок 58 г/л, креатинин 320 мкмоль/л. По УЗИ правая почка 13,2х6,8 см, левая – 14,0х7,0 см.
Задача № 2. 65-летняя женщина обследуется по поводу слабости и боли в ребрах и позвоночнике. Считает себя больной в течение 3 месяцев. 1 месяц назад лечилась по поводу перелома ребра. Объективно: бледная, отеки нижних конечностей. Периферические лимфоузлы не увеличены. Со стороны сердца и легких без патологии. Печень и селезенка не увеличены. Общий анализ крови: Hb - 110 г/л, Le - 7,8х109/л. СОЭ - 68 мм/ч. Общий анализ мочи: уд.вес - 1012, белок - 10 г/л, Le - 1-2 в п/зр, Er - 0-1 в п/зр. Общий белок сыворотки крови 60 г/л, альбумины 37,5%, глобулины: α1 – 8,5%, α2 – 18,2% β – 19,8%, γ – 16,0%. Миелограмма: сужение эритроидного ростка, плазматические клетки 20%.
Задача № 3. Женщина 26 лет обратилась с жалобами на внезапное развитие отеков на нижних конечностях и лице. 1 месяц назад перенесла мед. аборт на сроке 11 недель. Объективно: параорбитальные отеки, на скулах эритема, распространяющаяся к носогубной зоне, на нижних конечностях отеки до уровня колена. Температура тела 37ºС. Суставы внешне не изменены. Со стороны дыхательной и сердечно-сосудистой системы патологии не выявлено. Печень и селезенка не увеличены. Общий анализ крови: Hb 118 г/л, Le 2,6х109/л, СОЭ 40 мм/ч. Общий анализ мочи: уд. вес 1015, белок 5,06 г/л, Le 8-10 в п/зр, Er 10-12 в п/зр, гиалиновые цилиндры 4-5 в п/зр. Лейкоформула мочи: 60% лимфоцитов. Проба Зимницкого: ДД 400 мл, НД 350 мл, колебания уд. веса 1005-1018. СКФ 60 мл/мин. Анализы крови: мочевина 8,0 ммоль/л, креатинин 130 мкмоль/л, холестерин 7,5 ммоль/л. Общий белок 60 г/л, альбумины 38%, глобулины: α1 – 7%, α2 – 14% β – 12%, γ – 29%.
Задача № 4. Мужчина 46 лет, с 28 лет возникали рецидивирующие острые артриты до 3-4 раз в месяц, в 35 лет зафиксированы тофусы в области стоп, кистей, на ушных раковинах. В анализах мочи – снижение удельного веса. Год назад стали появляться отеки. В анализах мочи протеинурия до 2,5 г/л. Объективно: избыточного питания, параорбитальные отеки, отеки голеней до в/3. Внешне суставы не изменены. Тоны сердца приглушены. ЧСС 80 в мин. АД 140/100 мм рт. ст. Печень + 1,5 см от края реберной дуги, безболезненная. Общий анализ крови: Hb – 145 г/л, Tr – 850х109/л, Le – 6,8х109/л, СОЭ – 25 мм/ч. Общий анализ мочи: уд. вес 1010, белок 3,3 г/л, Le 2-3 в п/зр., ураты в большом кол-ве В анализах крови: билирубин 26,0-10,5-15,5 мкмоль/л, мочевая кислота 586 мкмоль/л, креатинин 151 мкмоль/л, мочевина 8,5 ммоль/л, общий белок 66 г/л, альбумины 39,2%, глобулины: α1 – 4,5%, α2 – 13,5% β – 18,1%, γ – 24,7%. СКФ 45 мл/мин.
Информационный блок Нефротический синдром (НС) – симптомокомплекс, сопровождающийся массивной протеинурией, гипоальбуминемией, отеками и гиперлипидемией. Основные причины нефротического синдрома представлены в таблице 1. Таблица 1. Этиология нефротического синдрома:
Обязательные признаки НС включают:
К дополнительным признакам относится нарушение обмена липопротеидов, фосфорно-кальциевого обмена, коагуляционные нарушения. Отеки при НС вызваны несколькими причинами, среди которых выделяют: 1) гипопротеинемию; 2) почечную задержку натрия; 3) системное нарушение сосудистой проницаемости. Таблица 2. Патогенетические варианты НС
Патогенез развития отеков при гиповолемическом варианте НС представлен на схеме: 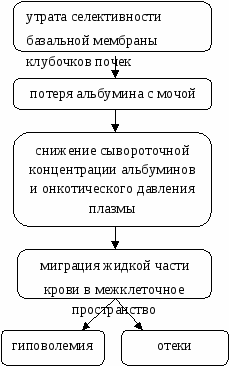 При гиперволемическом варианте, помимо снижения онкотического давления плазмы крови, на первое место, как причина отеков, выходит первичная задержка NaCl почками. Отеки могут развиваться постепенно или же бурно. Нефротические отеки рыхлые, легко перемещаются и оставляют ямку при надавливании пальцем. При больших отеках видны признаки дистрофии кожи и ее придатков: сухость, шелушение эпидермиса, ломкость и потускнение волос и ногтей. После схождения отеков заметна атрофия скелетной мускулатуры. По степени тяжести НС различают следующие формы:
Лечение: Этиотропная терапия зависит от основного заболевания (первичный нефрит, ВИЧ или гепатити-ассоциированная нефропатия, паранеопластический синдром и др.) Если причиной НС является гломерулонефрит, назначаются ГКС (преднизолон) в дозе 1 мг/кг ежедневно в течение двух месяцев с последующим медленным снижением дозы. При неэффективности ГКС назначают цитостатики (также в сочетании с небольшими дозами глюкокортикостероидов). При НС патогенетически обосновано применение антикоагулянтов, дезагрегантов. Лечение отеков Гипосолевая диета. При гиповолемическом варианте (альбумин менее 20 /л) начинают с введения онкотических плазмозамещающих растворов, лучше всего - альбумина. Петлевые диуретики, предпочтительно использовать те, которые не связываются с альбумином, например, торасемид. Фуросемид же связывается с альбумином, поэтому эффективность его в моче снижена. Антогоничты альдостерона (спироналактон) можно подключить при неэффективности монотерапии петлевыми диуретиками. Лечение гиперлипидемии – назначение статинов. Потенциальные осложнения НС делятся на:
Нефротический криз – резкое снижение объема циркулирующей крови, обусловленное падением онкотического давления, развивается у больных с нефротическим синдромом с выраженной гипоальбуминемией, проявляется выраженной артериальной гипотензией, вплоть до гиповолемического шока. Нефротический криз всегда развивается при уже сформировавшемся нефротическом синдроме (НС). Провоцирующие факторы:
В патогенезе нефротического криза одну из главных ролей играет локальная гиперпродукция кининов, что является специфичным именно для нефротического синдрома и не наблюдается при других «больших» нефрологических синдромах. С увеличением концентрации и продолжительности действия компонентов калликреин-кининовой системы связано появление таких характерных проявлений нефротического криза, как мигрирующая болезненная кожная эритема и боли в животе, которые являются предвестниками скорого наступления гиповолемического шока. Помимо повышения концентрации компонентов килликреин-кининовой системы, при гиповолемическом варианте нефротического синдрома наблюдается низкая активность ферментов, разрушающих кинины (кининаз, α1-антитрипсина, нейтральной эндопептидазы), активность которых также может быть подавлена при применении ингибиторов АПФ. Установлено, что повышение экскреции с мочой калликреина наблюдается у больных нефротическим синдромом как при нормальной, так и при повышенной активности ренина плазмы крови. Что говорит об исчезновении естественной физиологической взаимосвязи между активностью ренин-ангиотензин-альдостероновой и калликреин-кининовой систем [1]. Таким образом, артериальная гипотензия, возникающая и нарастающая при нефротическом кризе, обусловлена:
Клинические признаки нефротического криза:
Дифференциальная диагностика эритемы при нефротическом кризе и рожистом воспалении представлена в таблице 3. Таблица 3. Дифферениальная диагностика эритемы:
Лечение нефротического криза:
При гиповолемическом шоке требуется назначение вазопрессорных аминов: дофамин внутривенно капельно 0,5 % или 4 % раствор в ампулах по 5 мл разводят соответственно в 125 мл (0,2 мг/мл) или 400 мл (0,5 мг/мл) 5 % раствора глюкозы или изотонического раствора натрия хлорида. Начальная скорость введения составляет 1 – 5 мкг/кг в минуту (6 – 30 капель 0,02 % раствора, или 2 – 12 капель 0,05 % раствора). При необходимости скорость введения увеличивают до 10 – 25 мкг/кг в минуту (в среднем 18 мкг/кг в минуту). Инфузию производят непрерывно до нормализации АД. Амилоидоз – это группа заболеваний, характеризующихся отложением в органах и тканях аномального белка -фибриллярной структуры, что приводит к атрофии и склерозу паренхимы, недостаточности функции различных органов. Термин «амилоид» ввел в 1854 г. R. Virchow, который подробно изучил вещество, откладывающееся в тканях при так называемой сальной болезни у больных туберкулезом, сифилисом, актиномикозом, и счел его похожим на крахмал из-за характерной реакции с йодом. В дальнейшем была установлена белковая природа амилоида, но только через 100 лет после наблюдения R. Virchow с помощью электронного микроскопа (A.S. Cochen et al., 1959-1969) показана его фибриллярная структура с кросс--складчатой конформацией. В зависимости от особенностей этой структуры выделяют несколько типов амилоидоза. Классификация ВОЗ (1993) (табл. 4) построена по принципу специфичности основного фибриллярного белка амилоида для каждого типа амилоидоза. Во всех названиях типов амилоида первой буквой является прописная буква А, означающая слово «амилоид», за ней следует сокращенное обозначение конкретного фибриллярного белка амилоида: А – acquired (приобретенный), L – light chains (легкие цепи), TTR – transthyretin (транстиретин), β2-М – β2-микроглобулин соответственно. Таблица 4 Классификация системного амилоидоза (ВОЗ, 1993)
Таблица 5 Органы-мишени амилоидоза
АА-амилоидоз возникает как осложнение воспалительных или опухолевых заболеваний. В их числе:
Белок-предшественник SАА – острофазовый белок, продуцируемый в ответ на воспаление. SАА является -глобулином, близким по своим свойствам к СРБ. -глобулин синтезируется клетками разных типов (гепатоцитами, нейтрофилами, фибробластами) и поступает в кровеносное русло. Его синтез стимулируется ИЛ-1. Количество SAA повышается во много раз при воспалительных процессах и опухолях. На периферии, благодаря активности макрофагов, происходит протеолиз молекулы SАА и последующий синтез А-амилоида из образовавшихся обломков SАА. Депозиты АА-амилоида формируются во многих органах и системах: у большинства больных развивается амилоидоз почек, а также селезенки, кишечника, печени, нервной системы. Изменения функции почек, обусловленные отложением амилоида в клубочках, проявляются неселективной протеинурией с частым развитием нефротического синдрома и последующим формированием почечной недостаточности. Амилоидоз почек проходит 3 стадии: протеинурическую, нефротическую, азотемическую. Отличием нефротического синдрома при амилоидозе является гипергаммаглобулинемия, гиперфибриногенемия Б. Особенности ХПН – увеличенные или нормальные размеры почек, сохранение высокой протеинурии. Средний возраст заболевших – моложе 40 лет. АL-амилоидоз. Основным белком предшественником служат легкие цепи иммуноглобулинов, чаще (лямбда), реже (каппа) (3:1). АL-амилоидоз может развиваться как без признаков ранее или одновременно существующего заболевания, так и в сочетании с множественной миеломой и другой плазмоклеточной патологией. К плазмоклеточной дискразии относят болезни, при которых происходит избыточный синтез моноклональных иммуноглобулинов или их фрагментов вследствие пролиферации клона клеток В-лимфоцитов. К ним относятся доброкачественная моноклональная гаммапатия, множественная миелома, первичный АL-амилоидоз, макроглобулинемия Вальденстрема, болезнь тяжелых цепей, криоглобулинемия. К основным органам-мишеням относятся в первую очередь сердце (рестриктивная кардиопатия, кардиомегалия, нарушения ритма и проводимости, сердечная недостаточность, поражение клапанов и перикарда), ЖКТ (макроглоссия, синдром нарушенного всасывания, кровотечения), нервная система (периферическая нейропатия, вегетативная нейропатия, туннельный карпальный синдром), печень (гепато(сплено)мегалия, портальная гипертония, желтуха, гиперферментемия), а также почки. Дыхательная система вовлекается в процесс у 50 % больных. Среди клинических симптомов – охриплость голоса, диффузные изменения по типу фиброзирующего альвеолита с дыхательной недостаточностью и легочной гипертонией с развитием легочного сердца. Средний возраст заболевших при АL-амилоидозе – 65 лет. К ATTR-амилоидозу относится семейная амилоидная полинейропатия, кардиопатия, нефропатия с аутосомно-доминантным типом наследования и системный старческий амилоидоз (встречается на определенных территориях: Португалии, Швеции, Японии и т.д.). Белком-предшественником является транстиретин – транспортный белок для тироксина и ретинола, первично синтезируемый в печени. АTTR-амилоидоз является результатом мутации в гене, ответственном за синтез транстиретина, в котором происходит замена одной аминокислоты на другую. Обычно семейный амилоидоз проявляется только к середине жизни. Клинические проявления различаются при отдельных его вариантах, но наиболее часто отмечаются периферическая сенсорно-моторная нейропатия и нарушение автономных (вегетативных) функций. Кроме нервной системы могут быть вовлечены сердце, ЖКТ, почки. К АТТR-амилоидозу относится и системный старческий амилоидоз, при котором структура транстиретина не изменена. Является заболеванием исключительно пожилых людей (старше 70 лет). По клинике сходен с АL-амилоидозом, основными органами-мишенями являются сердце, сосуды, часто сочетается с атеросклерозом. А2М-амилоидоз – это диализный амилоидоз. Белок-предшественник – 2-микроглобулин, который не фильтруется через большинство диализных мембран и задерживается в организме. Основными органами-мишенями служат кости, периартикулярные ткани (переломы костей), синдром карпального канала – онемение и боль в первых трех пальцах кисти, распространяющаяся на предплечье, с последующей атрофией тенора из-за сдавления срединного нерва отложениями амилоида в области карпальной связки. Из приведенной классификации видно, что определенному биохимическому типу амилоидоза свойственна соответствующая клиническая картина заболевания. С другой стороны, определив клинический вариант амилоидоза, можно предположить, к какому типу амилоидоза относится данный случай. Так, при вторичном амилоидозе во всех случаях белок амилоида относится к АА-амилоиду. В связи с тем, что данная классификация учитывает происхождение амилоидоза и все клинические варианты заболевания, отпала необходимость в использовании прежней классификации амилоидоза, в основу которой был положен принцип наличия или отсутствия «причинного» фактора и выделялись идиопатический (первичный), наследственный (генетический), приобретенный (вторичный) и старческий амилоидозы. Патогенез амилоидоза до настоящего времени отстается недостаточно изученным. Наибольшим признанием пользуется иммунологическая теория патогенеза. Считается, что в условиях снижения функции Т-клеточного звена иммунитета и при наличии недостаточно изученного генетического дефекта макрофаги под влиянием ИЛ-1 вырабатывают предшественник амилоида. В условиях нарушенного выведения предшественника амилоида из организма из-за нарушенного катаболизма (в частности, в связи со снижением активности протеолитических ферментов), обусловленного генетическим дефектом в этой системе, происходит накопление предшественника амилоида в крови с последующим образованием фибрилл амилоида. Диагноз амилоидоза должен быть подтвержден морфологически при изучении материала, полученного при биопсии тканей. При АL-амилоидозе рекомендуют провести биопсию костного мозга с окраской на амилоид. Считают, что выявление амилоида в костном мозге с оценкой плазматических клеток дает представление сразу и о типе амилоидоза, так как амилоидоз костного мозга наиболее характерен для АL-амилоидоза. При подозрении на АL-амилоидоз также рекомендуют проведение аспирационной биопсии подкожно-жировой клетчатки из передней стенки живота с окраской на амилоид; положительный результат получают более чем в половине случаев. Информативным методом для диагностики амилоидоза является биопсия слизистой оболочки прямой кишки (50-70 % положительных результатов), десны (40-50 %), пораженного органа (90-100 %). Проводится окраска гистологических срезов конго красным с изучением в поляризационном свете, а также – тиофлавином Т, который обладает большей чувствительностью, но меньшей специфичностью. Поэтому для постановки диагноза применяют оба метода. Для установления типа амилоида проводят иммунопероксидазную реакцию с набором антисывороток к основным белкам амилоидных фибрилл. К достижениям последних лет относится метод сцинтиграфии с меченным 123J сывороточным Р-компонентом (SAP), предназначенный для оценки invivo распределения амилоида в организме. Метод особенно полезен для контроля за динамикой тканевых отложений амилоида в процессе лечения. Лечение амилоидоза направлено на уменьшение синтеза и доставки предшественников амилоида. Лечение прежде всего должно быть направлено на основное заболевание, что может остановить прогрессирование амилоидоза. Средства, используемые для воздействия непосредственно на амилоидоз, относятся в основном к разряду симптоматических, вместе с тем не исключается их умеренное действие на некоторые патогенетические механизмы. Характер фармакотерапии в существенной степени зависит от биохимического типа амилоидоза. С целью воздействия на АА-амилоидоз применяют димексид: 10-20 % раствор, разведенный в дистиллированной воде или соках, внутрь в суточной дозе 4-20 г, в течение 3-9 месяцев. Обладает противовоспалительным действием, обусловленным воздействием на гидроксильные радикалы, и прямым рассасывающим действием. К средствам, препятствующим образованию амилоида, относят унитиол. Унитиол (SH-содержащее соединение), «блокируя» реакционноспособные сульфгидрильные группы белковых субъединиц, тормозит агрегацию амилоидных фибрилл. Унитиол вводится в виде 5 % раствора внутримышечно по 5 мл 1 раз в сутки в течение 1 месяца, проводится несколько курсов с двухмесячным перерывом между ними. Колхицин используется как монотерапия при периодической болезни в дозе 1,8-2 мг/сут пожизненно. Он подавляет активность макрофагов и нейтрофилов, уменьшается выработка ИЛ-1, снижается синтез SАА, нарушается сборка амилоидных фибрилл на поверхностной мембране макрофагов. Лечение АL-амилоидоза направлено на уменьшение продукции предшественников – легких цепей иммуноглобулинов. Применяется наиболее хорошо апробированная схема полихимиотерапии: мелфалан из расчета 0,15 мг/кг массы тела в сочетании с преднизолоном 0,8 мг/кг/сут на протяжении 7 дней с перерывом 4-6 недель. Лечение должно быть длительным, не менее 1 года. Лечение застойной сердечной недостаточности при АL-ами-лоидозе сводится к назначению массивных доз мочегонных средств. Ни один метод лекарственной терапии не приводит к излечению от амилоидоза. Единственное, на что можно рассчитывать, – это замедление прогрессирования процесса. Заболевание неуклонно прогрессирует и заканчивается летально. Продолжительность жизни зависит от формы амилоидоза, сроков диагностики и степени вовлечения жизненно важных органов. При естественном течении АL-амилоидоза средний показатель выживаемости больных составляет 1-2 года. При АА-амилоидозе продолжительность жизни во многом зависит от основного заболевания и возможности его контролирования. Трансплантация почек – наиболее перспективный метод у больных с АА- и АL-амилоидозом с почечной недостаточностью. К сожалению, в трансплантированной почке развивается реамилоидоз, но для того, чтобы развилась почечная недостаточность, должно пройти не менее 15 лет (по данным литературы). |