|
|
Общие свойства катализаторов. Общая схема катализа. Сходство и р. Общие свойства катализаторов. Общая схема катализа. Сходство и различие процессов гомогенного и гетерогенного катализа
Федеральное государственное бюджетное общеобразовательное учреждение высшего образования
«Самарский государственный медицинский университет» Министерства здравоохранения Российской Федерации
Кафедра общей, бионеорганической и биоорганической химии
Реферат на тему:
Общие свойства катализаторов. Общая схема катализа. Сходство и
различие процессов гомогенного и гетерогенного катализа
Выполнила:
Студентка I курса лечебного факультета 122 группы
Прохорова А.С.
Проверил:
Д.б.н. профессор Агапов А.И.
Самара, 2020
Содержание:
1.Введение………………………………………………………………………....3
2. Сущность явления катализа и основные понятия каталитической химии…4
3. Автокатализ……………………………………………………………………4
4. Ферментативный катализ……………………………………………………....5
5.Основные типы катализаторов и механизмы каталитических реакций……..6
6. Гомогенный катализ…………………………………………………..………10
7.Гетерогенный катализ………………………………………………………....11
8. Заключение…………………………………………………………………….13
9. Список литературы…………………………………………………………....16
Введение:
Открытие явления катализа без сомнения следует отнести к величайшим достижениям химической науки, к важнейшему этапу в создании современной техники и эффективных технологий – цивилизации ХХ века. Явление катализа – основа существования живой клетки и, как полагают, могло иметь решающее значение в процессе возникновения жизни. Без каталитической химии сегодня трудно представить химическую промышленность, в которой более 90% всех процессов – каталитические процессы.
В задачу нашего реферата входит изложение современных представлений о сущности явления катализа и основных понятий каталитической химии. Также хотелось бы рассмотреть место каталитической химии в системе химических знаний.
Сущность явления катализа и основные понятия каталитической химии
Скорость химической реакции при данной температуре определяется скоростью образования активированного комплекса, которая, в свою очередь, зависит от величины энергии активации. Во многих химических реакциях в структуру активированного комплекса могут входить вещества, стехиометрически не являющиеся реагентами; очевидно, что в этом случае изменяется и величина энергии активации процесса. В случае наличия нескольких переходных состояний реакция будет идти в основном по пути с наименьшим активационным барьером.
Явление ускорения скорости химической реакции под действием веществ, непосредственно не участвующих в стехиометрическом уравнении реакции, называется катализом. Эти “сторонние” вещества, влияющие лишь на скорость реакции, называются катализаторами. Они не смещают химическое равновесие, и с его достижением могут быть выделены из реакционной смеси в химически неизменном виде. Их присутствие в равновесной системе сказывается лишь на тех термодинамических признаках системы, которые зависят от числа присутствующих в ней компонентов. Напомним, что в наиболее общей форме такое влияние должно учитываться через посредство активностей, связанных уравнением Гиббса-Дюгема, но в этом почти не бывает необходимости.
Различают положительный и отрицательный катализ (соответственно увеличение и уменьшение скорости реакции), хотя часто под термином "катализ" подразумевают только положительный катализ; отрицательный катализ называют ингибированием.
Автокатализ
Автокатализ – процесс каталитического ускорения химической реакции одним из её продуктов. В качестве примера можно привести катализируемую ионами водорода реакцию гидролиза сложных эфиров. Образующаяся при гидролизе кислота диссоциирует с образованием протонов, которые ускоряют реакцию гидролиза. Особенность автокаталитической реакции состоит в том, что данная реакция протекает с постоянным возрастанием концентрации катализатора. Поэтому в начальный период реакции скорость её возрастает, а на последующих стадиях в результате убыли концентрации реагентов скорость начинает уменьшаться; кинетическая кривая продукта автокаталитической реакции имеет характерный S-образный вид.
Ферментативный катализ
Ферментативный катализ – каталитические реакции, протекающие с участием ферментов – биологических катализаторов белковой природы. Ферментативный катализ имеет две характерные особенности:
1. Высокая активность, на несколько порядков превышающая активность неорганических катализаторов, что объясняется очень значительным снижением энергии активации процесса ферментами. Так, константа скорости реакции разложения перекиси водорода, катализируемой ионами Fе2+, составляет 56 с-1; константа скорости этой же реакции, катализируемой ферментом каталазой, равна 3.5·107, т.е. реакция в присутствии фермента протекает в миллион раз быстрее (энергии активации процессов составляют соответственно 42 и 7.1 кДж/моль). Константы скорости гидролиза мочевины в присутствии кислоты и уреазы различаются на тринадцать порядков, составляя 7.4·10-7 и 5·106 с-1 (величина энергии активации составляет соответственно 103 и 28 кДж/моль).
2. Высокая специфичность. Например, амилаза катализирует процесс расщепления крахмала, представляющего собой цепь одинаковых глюкозных звеньев, но не катализирует гидролиз сахарозы, молекула которой составлена из глюкозного и фруктозного фрагментов.
Согласно общепринятым представлениям о механизме ферментативного катализа, субстрат S и фермент F находятся в равновесии с очень быстро образующимся фермент-субстратным комплексом FS, который сравнительно медленно распадается на продукт реакции P с выделением свободного фермента; т.о., стадия распада фермент-субстратного комплекса на продукты реакции является скоростьопределяющей (лимитирующей).
Исследование зависимости скорости ферментативной реакции от концентрации субстрата при неизменной концентрации фермента показали, что с увеличением концентрации субстрата скорость реакции сначала увеличивается, а затем перестает изменяться и зависимость скорости реакции от концентрации субстрата описывается следующим уравнением:
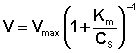
Здесь Кm – константа Михаэлиса, численно равная концентрации субстрата при V = ½Vmax. Константа Михаэлиса служит мерой сродства между субстратом и ферментом: чем меньше Кm, тем больше их способность к образованию фермент-субстратного комплекса.
Характерной особенностью действия ферментов является также высокая чувствительность активности ферментов к внешним условиям – рН среды и температуре. Ферменты активны лишь в достаточно узком интервале рН и температуры, причем для ферментов характерно наличие в этом интервале максимума активности при некотором оптимальном значении рН или температуры; по обе стороны от этого значения активность ферментов быстро снижается.
Основные типы катализаторов и механизмы каталитических реакций.
Катализатор - вещество переводящее систему реагентов в реакционноспособное состояние путём образования с реагентами лабильного каталитического комплекса.
Практически все катализаторы можно разделить на 5 типов, учитывая особенности их строения и механизма катализа:
1. Кислоты и основания (гомогенные и гетерогенные катализаторы) – протонные кислоты Бренстеда (НА) в водных и неводных средах, апротонные кислоты Льюиса–Усановича (BF3 , RI), протонные и апротонные центры твердых оксидов (γ-Al2 O3 ,Al2O3 –SiO2 , цеолиты), любые типы оснований (в том числе твердые – MgO, CaCO3 , анионообменные смолы).
2.Комплексы металлов (гомогенные и гетерогенные катализаторы) – MLn , Mm Ln .
3.Твердые соединения металлов типа Mm Эn , где Э = O, S, Se, Te, As, P, C, N, Si, B, H, – гетерогенные катализаторы.
4.Металлические катализаторы (гетерогенные) – нанесенные на инертных носителях (Pt/Al2 O3 ) или массивные металлы и сплавы.
5.Ферменты (гомогенные и гетерогенные).
Рассмотрим особенности механизма действия этих групп катализаторов.
Кислотно-основной катализ относится к очень распространенному и к наиболее изученному типу катализа. В катализе протонными кислотами Бренстеда (НА) субстрат реакции (реагент) выступает в качестве основания и первой стадией является протонирование реагента. Протонированный реагент (B) переходит в более реакционно-способное состояние и превращается далее через одно или несколько промежуточных соединений. В результате первой стадии переноса протона на олефин образуется новая кислота (апротонная) – ион карбения. Эта частица содержит положительно заряженный атом углерода (карбокатион) с вакантной орбиталью.
Такой катион (R+ – кислота по Льюису) реагирует со второй молекулой олефина как с основанием и вновь образует ион карбения. Этот новый катион может отщепить кислоту-катализатор, и тогда мы получим продукт каталитической димеризации олефина. Этот же катион может прореагировать последовательно с несколькими молекулами олефина, что приведет к процессу полимеризации олефина.
Если в системе присутствует соответствующий алкан, то ион карбения, отщепляя от него гидридион (H− ), превратится в изопарафин, а из алкана образуется новый ион карбения. В этом случае мы получим продукт алкилирования парафина олефином, причем образовавшийся на первой стадии ион карбения и будет катализатором процесса алкилирования.
Любое органическое и неорганическое соединение может выступать в роли основания, однако чем слабее основность соединения, тем более сильная кислота требуется для его протонирования. Так, очень сильные протонные кислоты (“суперкислоты”, “магические” кислоты), образующиеся в системах HF–SbF5 , ( H + [ SbF - 6 ]), и HSO3 F–SbF5 (H2 SO 3 F + [ SbF 5 ( OSO 2 F )− ]) протонируют в мягких условиях даже парафины . Так, метан образует ион карбония (ион метония).
Металлокомплексный катализ – быстроразвивающаяся область каталитической химии. Более 50 крупнотоннажных промышленных процессов используют гомогенные или гетерогенные металлокомплексные катализаторы. Химия комплексных соединений (координационная химия) и химия металлоорганических соединений являются основой этой области каталитической химии.
Координационная химия после создания теории комплексных соединений А. Вернером (1893 – 1905гг.) прошла большой путь и стала, по существу, языком неорганической и металлоорганической химии. Установлено, что простых соединений, в которых число двухэлектронных связей соответствует степени окисления металла комплексообразователя, практически не существует.
Так, например, молекула HgCl2 существует в виде линейной молекулы Cl–Hg–Cl только в парах при высоких температурах (>100°С). В твердой фазе и в растворах как соли, так и гидроксиды тяжелых металлов существуют в виде координационных соединений, в которых атом металла окружен различными группами (лигандами), например октаэдр HgCl2 (H2 O)4 в воде. Важнейшую роль в развитии металлокомплексного органического катализа сыграло открытие реакций окисления олефинов в растворах солей палладия, о котором мы говорили выше, и изучение их механизма (И.И. Моисеев, П. Генри).
Гетерогенный катализ металлами и оксидами металлов . Каталитические реакции на поверхности так же, как и реакции на поверхности электродов (электрохимия) и фотохимические процессы в тонких пленках (фотография), относятся к особой области химии – химии поверхности.
К реакциям на поверхности переходных металлов, оксидов металлов и других металлсодержащих соединений применимы все представления координационной и металлоорганической (в случае органических реакций) химии. На поверхности металлов образуются первичные комплексы реагентов с атомом или группой атомов поверхности и продукты их превращений.
Адсорбированные молекулы изменяются так же сильно, как и в процессах комплексообразования (особенно с кластерами металлов). Помимо химических проявлений, в гетерогенном катализе следует учитывать наличие объема твердого тела (большого количества атомов) и ряд особенностей поверхности.
Биокатализаторы – ферменты . Ферменты – это молекулы белка, которые в большинстве случаев растворимы в воде, но иногда находятся в коллоидном (микрогетерогенном) состоянии. Активные центры фермента формируются в результате стягивания в одну область пространства различных функциональных групп, принадлежащих различным аминокислотным фрагментам молекулы белка (COOH,
OH,
NH2,
SH,
имидазол и др.).
Активный центр располагается в виде щели в глобуле белковой молекулы. В некоторых ферментах и коферментах (молекулах, выполняющих роль специфических реагентов, регенерируемых в ходе реакций с другими ферментами) присутствуют ионы металлов (Fe, Cu, Zn, Mo, V, Co и др.), окруженные макролигандами.
Одним из таких Co-содержащих коферментов является витамин В12 . Наряду с высокой частотой оборотов фермента (А) при относительно небольшом времени жизни ферментам свойственна высокая специфичность по отношению к определенному субстрату и высокая специфичность к типу реакции.
Высокие активность и селективность действия фермента достигаются благодаря высокой степени организации активного центра и некоторым особенностям действия белковых катализаторов:
а) первичное связывание реагента в активном центре весьма специфично;
б) молекула реагента, попав в активный центр, вызывает изменение структуры белковой молекулы и подстраивает “под себя” геометрию активного центра так, чтобы дальнейшее превращение было наиболее выгодным (протекало с большей скоростью);
в) в активном центре на молекулу реагента обычно синхронно действуют несколько активных групп (кислотных, основных, нуклеофильных каталитических центров), что напоминает действие сварочных роботов на конвейере сборки автомобилей.
Роль объединения различных групп в одном катализаторе и синхронного их действия давно известна и в химическом кислотно-основном катализе.
Необходимо отметить, что наличие катализатора не влияет на величину изменения термодинамического потенциала в результате процесса и, следовательно, никакой катализатор не может сделать возможным самопроизвольное протекание термодинамически невозможного процесса (процесса, ΔG (ΔF) которого больше нуля). Катализатор не изменяет величину константы равновесия для обратимых реакций; влияние катализатора в этом случае заключается только в ускорении достижения равновесного состояния.
Гомогенный катализ
Гомогенный катализ – каталитические реакции, в которых реагенты и катализатор находятся в одной фазе. В случае гомогенно-каталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты.
Рассмотрим некоторую реакцию:
А + В ––> С
В присутствии катализатора осуществляются две быстро протекающие стадии, в результате которых образуются частицы промежуточного соединения АК и затем (через активированный комплекс АВК#) конечный продукт реакции с регенерацией катализатора:
А + К ––> АК
АК + В ––> С + К
Примером такого процесса может служить реакция разложения ацетальдегида, энергия активации которой EA = 190 кДж/моль:
СН3СНО ––> СН4 + СО
В присутствии паров йода этот процесс протекает в две стадии:
СН3СНО + I2 ––> СН3I + НI + СО
СН3I + НI ––> СН4 + I2
Уменьшение энергии активации этой реакции в присутствии катализатора составляет 54 кДж/моль; константа скорости реакции при этом увеличивается приблизительно в 105 раз. Наиболее распространенным типом гомогенного катализа является кислотный катализ, при котором в роли катализатора выступают ионы водорода Н+.
Гетерогеннвй катализ.
Гетерогенный катализ – каталитические реакции, идущие на поверхности раздела фаз, образуемых катализатором и реагирующими веществами. Механизм гетерогенно-каталитических процессов значительно более сложен, чем в случае гомогенного катализа. В каждой гетерогенно-каталитической реакции можно выделить как минимум шесть стадий:
1. Диффузия исходных веществ к поверхности катализатора.
2. Адсорбция исходных веществ на поверхности с образованием некоторого промежуточного соединения:
А + В + К ––> АВК
3. Активация адсорбированного состояния (необходимая для этого энергия есть истинная энергия активации процесса):
АВК ––> АВК#
4. Распад активированного комплекса с образованием адсорбированных продуктов реакции:
АВК# ––> СDК
5. Десорбция продуктов реакции с поверхности катализатора.
СDК ––> С + D + К
6. Диффузия продуктов реакции от поверхности катализатора.
Специфической особенностью гетерокаталитических процессов является способность катализатора к промотированию и отравлению.
Промотирование – увеличение активности катализатора в присутствии веществ, которые сами не являются катализаторами данного процесса (промоторов). Например, для катализируемой металлическим никелем реакции
СО + Н2 ––> СН4 + Н2О
введение в никелевый катализатор небольшой примеси церия приводит к резкому возрастанию активности катализатора.
Отравление – резкое снижение активности катализатора в присутствии некоторых веществ (т. н. каталитических ядов). Например, для реакции синтеза аммиака (катализатор – губчатое железо), присутствие в реакционной смеси соединений кислорода или серы вызывает резкое снижение активности железного катализатора; в то же время способность катализатора адсорбировать исходные вещества снижается очень незначительно.
Для объяснения этих особенностей гетерогенно-каталитических процессов Г. Тэйлором было высказано следующее предположение: каталитически активной является не вся поверхность катализатора, а лишь некоторые её участки – т.н. активные центры, которыми могут являться различные дефекты кристаллической структуры катализатора (например, выступы либо впадины на поверхности катализатора). В настоящее время нет единой теории гетерогенного катализа. Для металлических катализаторов была разработана теория мультиплетов. Основные положения мультиплетной теории состоят в следующем:
1. Активный центр катализатора представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности катализатора в геометрическом соответствии со строением молекулы, претерпевающей превращение.
2. При адсорбции реагирующих молекул на активном центре образуется мультиплетный комплекс, в результате чего происходит перераспределение связей, приводящее к образованию продуктов реакции.
Теорию мультиплетов называют иногда теорией геометрического подобия активного центра и реагирующих молекул. Для различных реакций число адсорбционных центров (каждый из которых отождествляется с атомом металла) в активном центре различно – 2, 3, 4 и т.д. Подобные активные центры называются соответственно дублет, триплет, квадруплет и т.д. (в общем случае мультиплет, чему и обязана теория своим названием).
Заключение:
Итак, катализ – это ускорение или возбуждение химических реакций в присутствии веществ – катализаторов, многократно вступающих в промежуточное химическое взаимодействие с участниками реакции и восстанавливающих после каждого цикла свой первоначальный химический состав. Это определение не содержит никаких указаний на причину, по которой такое промежуточное взаимодействие между катализатором и реагентами вызывает ускорение химической реакции, но лишь описывает новый путь, по которому идет эта реакция с участием катализатора.
Катализ занимает особое место как в системе наших знаний о веществах и их превращениях, так и в практической деятельности человека. Он лежит в основе существования растительного и животного мира, обеспечивая с помощью ферментов функционирование живых систем. В истории нашей цивилизации катализ не раз становился решающим фактором технического прогресса. Достаточно назвать лишь три промышленных каталитических процесса: синтез аммиака, крекинг нефти и полимеризация олефинов, чтобы убедиться в этом.
Все сказанное в достаточной мере характеризует ту важную роль, которую играет катализ в сфере научной и практической деятельности современного человека. Существует, однако, одна специфическая особенность катализа, которая делает постановку вопроса, вынесенного в заголовок статьи, хотя и несколько неожиданной, но вполне оправданной. В отличие от других разделов химии в катализе пока не существует общей теории, способной заранее предсказать, будет ли данное вещество ускорять какую-либо реакцию.
Широко известные теории катализа такие, как мультиплетная теория Баландина, теория активных ансамблей Кобозева, теория катализа на полупроводниках Доудена и многие другие теоретические концепции являются не общими, а частными моделями, относящимися к сравнительно узкому кругу каталитических систем. Но и эти частные теории лишь объясняют опытные факты.
Отсутствие научной теории, способной предсказать каталитические свойства веществ, как раз и породило неоднократно встречавшееся в специальной литературе суждение о катализе скорее как об искусстве, чем науке. Полуторавековая история катализа как самостоятельного раздела химии показывает, что все известные катализаторы были открыты либо случайно, либо интуитивно, эмпирически.
Объективную причину столь необычной в современной науке ситуации следует, по-видимому, искать в самой природе катализа и поразительном разнообразии каталитических процессов. Даже сейчас, когда новейшие экспериментальные методы позволяют подойти к изучению процессов катализа, а также самих катализаторов на атомно-молекулярном уровне и в масштабе реального времени, Природа не открывает тайну, как она это делает.
Список литературы:
1. Боресков Г.К. Гетерогенный катализ. – М.: Наука, 1999. – 304 с.
2. Боресков Г.К. Катализ. Избранные труды. – Новосибирск: Наука, 2004.
3. Гейтс Б., Кетцир Дж., Шуйт Г. Химия каталитических процессов. – М.: Мир, 2000.
4. Кузнецов В.И. Развитие учения о катализе. – М.: Наука, 1994.
|
|
|
 Скачать 39.12 Kb.
Скачать 39.12 Kb.