Презентация. материал по ЦГ. Основы цитогенетики Цитогенетика
 Скачать 340.5 Kb. Скачать 340.5 Kb.
|

|
Основы цитогенетики Цитогенетика – раздел клинической генетики, изучающий клеточные основы наследственности и изменчивости. Медицинская цитогенетика – изучает связь изменчивости хромосом с патологией у человека. Термин цитогенетика ввел Саттон в 1903 г. Основными задачами цитогенетики являются: ● определение состояния хромосомного набора человека в норме; ● сопоставление клиники и различных видов хромосомных аберраций; ● разработка и использование методов цитогенетической диагностики хромосомных болезней (ХБ); ● разработка методов ранней диагностики и профилактики хромосомных болезней; ● совершенствование медико-генетического консультирования при ХБ ● создание регистров хромосомных болезней. Хромосома (от греч. chromo – цвет, soma – тело) – материальный носитель наследственной информации, передаваемой от поколения к поколению, входящий в состав клеточных ядер и имеющий хроматиновый состав. Во втором десятилетии XX века Т.Г. Морган и его школа сформулировали основные положения «хромосомной теории наследственности»: гены расположены в хромосомах, каждая хромосома представляет собой группу сцепления генов (обеспечивает совместное наследование признаков). Число групп сцепления у каждого вида равно гаплоидному числу хромосом. Число генов пропорционально длине хромосом; каждый ген в хромосоме занимает определенное место (локус). Гены расположены в хромосоме линейно. Кроме них выделяют «прыгающие гены», или транспозоны, которые могут перемещаться по геному; между гомологичными хромосомами может происходить обмен аллельными генами. В гомологичных (одинаковых) хромосомах по происхождению одна – отцовская, другая – материнская. Расположенные в них друг против друга локусы называются идентичными, и в таких локусах расположены аллельные гены; расстояние между генами в хромосоме пропорционально проценту кроссинговера между ними. Кроссинговер – процесс гомологичной рекомбинации, протекающий в первом мейотическом деление в ходе оогенеза и сперматогенеза. Хромосома – это уровень организации наследственного материала, представленного в виде хроматина или хроматинового комплекса. Хроматин – вещество, составляющее хромосому, представляет собой двойную нить ДНК связанную с белками (дезоксирибонуклеопротеидный комплекс). Белки, входящие в состав хроматина разнообразны. Их разделяют на две группы: гистоны и негистоновые белки. Гистоны характерны только для эукариотических клеток, они осуществляют первые этапы упаковки ДНК. Их взаимодействие с ДНК происходит за счет ионных связей. Это глобулярные белки, представленные 5-7-ю видами молекул. Наиболее известны следующие классы гистонов: Н1, Н2А, Н2В, НЗ, Н4. Их основные свойства определяются относительно высоким содержанием аминокислот, лизина и аргинина. Первый этап упаковки ДНК в хроматине осуществляется с помощью нуклеосом – это дисковидные частицы диаметром 10 нм. Молекула ДНК накручивается на поверхность множества белковых частиц, каждый раз делая 1,75 оборота вокруг сердцевины. Сердцевина нуклеосомы всегда консервативна и содержит 8 молекул – по 2 молекулы гистонов Н4, НЗ, Н2А, Н2В. По поверхности сердцевины располагается участок ДНК из 146 нуклеотидных пар. Небольшой участок ДНК между нуклеосомами остается несвязанным с сердцевиной, он называется линкером. Последовательность из нуклеосом – нуклеофиламент (nucleofilament), или нуклеосомная нить. Общий вид хроматина, представленного молекулой ДНК, упакованной с помощью нуклеосомных структур, можно сравнить с «бусами на нитке». Первый нуклеосомный уровень компактизации укорачивает молекулу ДНК в 6-7 раз. В следующий этап упаковки нуклеосомная структура хроматина вовлекается с помощью гистона Н1, который связывается с линкерной частью ДНК и поверхностью нуклеосомы. Возникает упорядоченная структура спирального типа, которую называют соленоидом. Его диаметр 25-50 нм – спираль более высокого порядка. Под электронным микроскопом соленоид выявляется в виде фибрилл хроматина. Фибриллы ДНК попарно скручиваясь, образуют хромонемы (от греч. nema-струна), которая входит в комплекс более высокого порядка полухроматиды (спирально закручены). Перед началом деления клеточного ядра хромосома спирализуется, образуя при помощи белка H1 более толстую хроматиновую нить или хроматиду (chromatin fiber), диаметром 30 нм. Дальнейшая суперспирализация хроматина приводит к образованию пары хроматид. Пара хроматид образует хромосому. Толщина хромосомы (диаметр - 1400 нм) на стадии метафазы позволяет её увидеть в световой микроскоп. 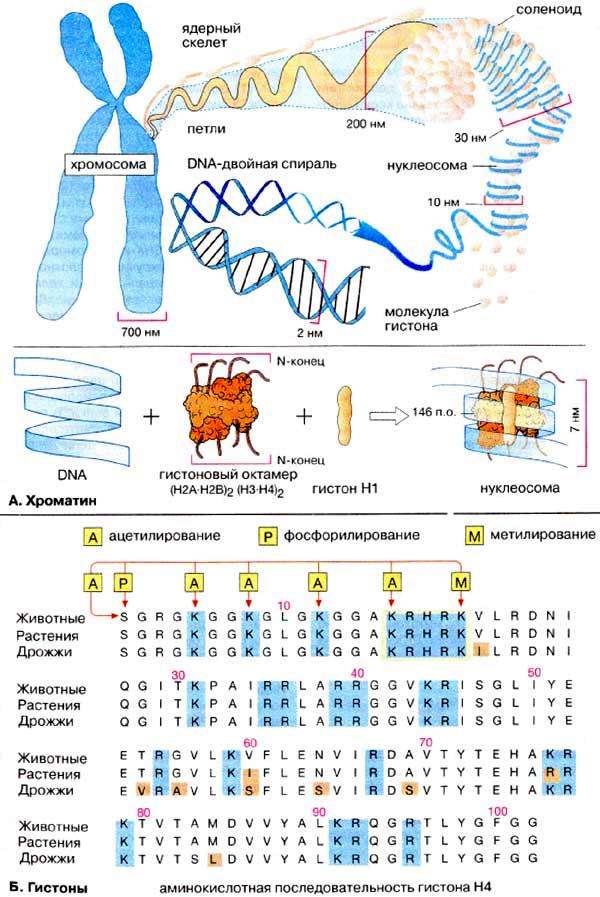 Рис. 4. Упаковка ДНК в хроматине. Хроматиды соединяются между собой в районе первичной перетяжки или центромеры. Центромеры выполняют в хромосомах очень важные функции, они соединяют две сестринские хроматиды, играют важную роль в организации веретена деления (на ней находится кинетохор, к которому присоединяются нити веретена при делении). Внутри центромеры выявляются уникальные последовательности, которые, вероятно, несут информацию о расхождении хромосом к противоположным полюсам клетки. Благодаря первичной перетяжке хромосома делится на плечи, короткое (p) и длинное (q). Вторичные перетяжки у человека локализуются в разных точках по длине хромосом - 13, 14, 15, 21 и 22. Сателлит(спутник) — это округлое или удлинённое тельце, отделённое от основной части хромосомы тонкой хроматиновой нитью, по диаметру равный или меньший хромосоме. Хромосомы, обладающие спутником – SAT-хромосомы. В зависимости от размера, положения центромеры и длины плеча выделяют три типа хромосом: ● медианный (метацентрический) – центромера расположена в середине хромосомы и оба плеча равны. ● субмедианный (субметацентрический) – центромера расположена ближе к одному из концов, плечи хромосомы не равны: ● субтерминальный (акроцентрический) – центромера расположена на конце хромомсомы. Для измерения длины хромосомы был предложен центромерный индекс – доля длины короткого плеча длине всей хромосомы, принятой за 100%. Если центромерный индекс равен 50%, то это метацентрическая хромосома, если меньше 50% - субметацентрическая хромосома, а если центромера расположена близко к концу хромосомы – то акроцентрическая. 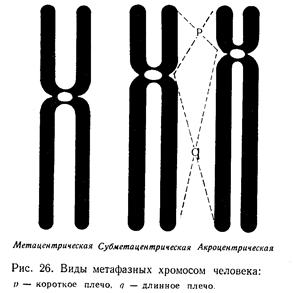 Рис.5. Типы хромосом человека. Концевая часть хромосомы называется теломерой (от греч. telos - конец и meros - часть). Она состоит из линейной хромосомной ДНК, состоящей из коротких тандемных повторов, образующей конститутивный теломерный гетерохроматин. Теломера выполняет защитную функцию, предотвращает слипание хромосом своими концами, стабилизирует хромосомы, защищая их от деградации клеточными нуклеазами, препятствует образованию дицентриков. Она играет роль в создании специфической архитектуры ядра клетки. Благодаря теломере, происходит полное завершение редупликации хромосом при подготовке клетки к делению. В каждом цикле деления теломеры клетки укорачиваются - феномен концевой недорепликации (важнейший фактор биологического старения). Специальный фермент - теломераза при помощи собственной РНК-матрицы достраивает теломерные повторы и удлиняет теломеры. В большинстве дифференцированных клеток теломераза заблокирована, активна в стволовых и половых клетках. В 2009 г. Нобелевская премия по физиологии и медицине была присуждена за открытие защитных механизмов хромосом от концевой недорепликации с помощью теломер и теломеразы Элизабет Блекберн (Elizabeth Blackburn), Кэрол Грейдер (Carol Greider) и Джек Шостак (Jack Szostack). На различных участках одной и той же хромосомы спирализация, компактность ее основных элементов неодинакова. Выделяют активные районы, которые содержат гены, контролирующие развитие признаков организма; в интерфазных ядрах это деконденсирующиеся, слабоокрашивающиеся нитчатые структуры. Хроматин, представлен здесь в активной форме, в нем происходит транскрипция – синтез всех типов молекул РНК. Эти районы называют эухроматические. Соответственно, эухроматинизация – деспирализация участков хромосом в интерфазном ядре, сопровождающаяся их генетической активацией. Также выделяют неактивные участки, они отличаются высокой степенью спирализации, сохраняются на протяжении всего митотического цикла, интенсивно окрашиваются, не содержат уникальных генов, и получили название гетерохроматические. Гетерохроматин делят на структурный и факультативный. Факультативный гетерохроматин (ФГ) содержит кодирующую ДНК, неактивные в данной клетке гены. Его участки присутствуют только в одной из гомологичных хромосом. Типичный пример факультативного гетерохроматина - неактивная половая хромосома (в интерфазе как тельце Барра). ФГ обуславливает «молчание» тканеспецифичных генов. Конститутивный (структурный) гетерохроматин содержится в обеих гомологичных хромосомах, локализован в центромере, теломерах, ядрышковом организаторе, в нем нет уникальных генов. Его ДНК преимущественно сателлитная, состоящая из тандемных повторов. Он выполняет структурную и регуляторную функцию, служит защитным экраном для эухроматина. Термины эухроматин и гетерохроматин предложил Хайц в 1928 г. Хромосомы клетки можно увидеть только во время ее деления – при митозе (форма деления соматических клеток) и мейозе (форма деления половых клеток), их называют метафазные пластинки. Номенклатура хромосом В г. Денвере (США) в 1960 г. была проведена первая международная научная конференция цитогенетиков, на которой предложена номенклатура хромосом человека в зависимости от их морфологической характеристики, учитывающей размеры, форму и положение центромеры, положившая основу для всех последующих номенклатур. Согласно Денверской классификации все аутосомы получили порядковые номера и были разделены на 7 групп: Группа А (1-3) - большие метацентрические, Группа В (4,5) - крупные субметацентрические, Группа С (6-12) - средние субметацентрические, Группа D (13-15) - большие акроцентрические, Группа Е (16-18) – короткие субметацентрические, Группа F (19,20) - малые метацентрические, Группа G (21,22) – малые акроцентрические, Х-хромосома - ближе по своей морфологии к группе С, Y-хромосома - ближе к группе G. Затем были уточнения данной классификации хромосом в 1963г. в Лондоне и в 1966г. в Чикаго. В начале 70-х годов ХХ века для точной идентификации хромосом были разработаны различные методы дифференциального окрашивания. В 1971 г. в Париже на IV международном конгрессе по генетике человека была согласована единая система идентификации хромосом человека, учитывавшая дифференцировку хромосом по длине. Каждая хромосома набора человека при дифференциальной окраске характеризуется уникальным для нее сочетанием темно окрашенных сегментов или полос, «бэндов» (от англ. band – полоска), чередующихся с неокрашенными участками или светлыми сегментами. Такое специфическое для данной хромосомы сочетание сегментов позволяет четко ее идентифицировать и отличить от других хромосом набора. В пределах короткого (p) и длинного (q) плеча каждой хромосомы выделяют ряд четко идентифицируемых областей или регионов (от англ. region), которые нумеруются арабскими цифрами от центромеры (cen) к теломерному участку (tel) или терминальному (ter) концу хромосомы. Каждая область включает определенное число сегментов, нумерация которых (второй арабской цифрой) также идет от центромерного к теломерному участку. Например, обозначение хромосомного сегмента 2q34 означает следующее: 2 – хромосома 2, q – длинное плечо, 3 – регион, 4 – сегмент. В 1978 г. издан унифицированный вариант номенклатуры хромосом человека (обобщенные данные) – ISCN – «An International System for Human Cytogenetic Nomenclature (1978)». В середине 70-х годов ХХ века были разработаны методы получения хромосом высокого разрешения. В результате то, что раньше представлялось единой светлой или темной полосой, приобретает собственную специфическую исчерченность. Проявляются так называемые субсегменты. Например, если сегмент 1p31 подразделяется на 3 разных субсегмента, то они обозначаются как 1p31.1, 1p31.2, 1p31.3, при этом субсегмент 1p31.1 является проксимальным, а 1p31.3 –дистальным по отношению к центромере. Все это нашло отражение в международном соглашении в 1981 г. в Париже, которое было опубликовано под названием «Международная система номенклатуры цитогенетики человека – исчерченность высокого разрешения» или «ISCN 1981». В 1985г. в г. Иерусалиме принята цитогенетическая номенклатура, учитывающая приобретенные конституциональные хромосомные нарушения в опухолевых клетках - «ISCN 1985». В 1991 г. в Берлине издано «Руководство по цитогенетике рака» «ISCN 1991». В 1995г. в Вашингтоне принят окончательный документ «ISCN 1995», в котором отражены рекомендации по онкологической цитогенетике и достижения, полученные в ходе использования FISH-методов диагностики. Номенклатура была модернизирована и дополнена, в том числе сведениями о способах описания результатов сравнительной геномной гибридизации в 2005 году - «ISCN 2005». Кариотип – систематизированный набор хромосом ядра клетки с его количественными и качественными характеристиками («лицо вида»). Нормальный кариотип человека – 46,ХХ (женщина) и 46,ХУ (мужчина), т.е. диплоидный (двойной) набор хромосом, или 22 пары аутосом (хромосомы тела) и 1 пара гоносом (половые хромосомы). Впервые термин был введен в 1924 г. Г.А. Левитским. Диплоидный набор – сумма 2 гаплоидных наборов – 2n (яйцеклетка и сперматозоид), является нормой для соматических клеток. Он приводит к взаимосвязанной работе двух аллелей каждого гена и обеспечивает внутривидовое разнообразие. У женщин гоносомы представлены двумя гомологичными (идентичными) Х-хромосомами, так как в процессе оогенеза образуется один тип яйцеклеток, содержащих Х-хромосому. У мужчин гоносомы представлены одной Х-хромосомой и одной У-хромосомой, так как в процессе сперматогенеза образуются два типа сперматозоидов, имеющих X-хромосому, либо У-хромосому (их соотношение примерно 1:1). Определение пола человека происходит в момент оплодотворения яйцеклетки сперматозоидом. Оно целиком зависит от того, какой это сперматозоид: если с Х-хромосомой, то будет женский организм, если с У-хромосомой, то мужской организм. Согласно современной номенклатуре хромосом блоки информации в формуле кариотипа следующие: ● общее число хромосом; ● перечисление половых хромосом; ● блоки числовых аномалий (каждая аномалия – свой блок); ● блоки структурных аномалий (каждая аномалия – свой блок); ● [ , ] – разделяет только блоки информации; ● [ ; ] – отделяет информацию о различных хромосомах в пределах одного блока; ● [ . ] – отделяет обозначение субсегмента; ● [ : ] – обозначает точки разрывов и соединений в развёрнутой формуле. Также указываются индивидуальные особенности хромосомы (естественный полиморфизм), родительское происхождение гомологичных хромосом. 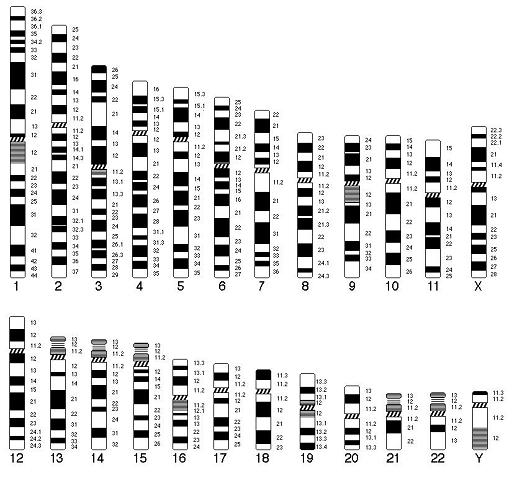 Рис. 6. Схематическое изображение хромосом при G-окрашивании в соответствии с международной классификацией. Патологический кариотип – вариант кариотипа, являющегося причиной различных нарушений фенотипа и функционирования организма, может быть обусловлен хромосомными и геномными мутациями (см. главу «Изменчивость»). Контрольно-обучающие вопросы 1. Перечислите основные задачи клинической цитогенетики. 2.Сформулируйте основные положения «хромосомной теории наследственности». 3. Какие группы белков входят в состав хроматина? 4. Какие функции выполняют в хромосомах центромеры? 5. Какую роль играют теломеры в феномене концевой недорепликации? 6. Перечислите типы хромосом у человека. 7. Какие принципы положены в основу Денверской классификации хромосом? 8. Назовите символы, используемые для обозначения хромосомного сегмента. 9. Дайте определение термина «кариотип». 10.Какие районы выделяют в хромосоме в зависимости от степени ее спирализации? Хромосомные мутации Хромосомные мутации (перестройки, аберрации) – это мутации, связанные с изменением структуры хромосом. Они не касаются количества хромосом в наборе. Структурные нарушения являются результатом одного и более разрывов в одной хромосоме (внутрихромосомные перестройки) или двух и более разрывов в разных хромосомах (межхромосомные перестройки). В любом случае происходит разрыв сахаро-фосфатных связей ДНК. Воссоединение разорванных концов осуществляется за счет ферментов репарации. Если концы хромосомных фрагментов воссоединяются как прежде, то хромосома и клетка остаются интактными. Если происходит потеря хромосомного материала или воссоединение хромосомных фрагментов в точках разрывов других хромосом, то возникают различные структурные хромосомные перестройки. В зависимости от воздействия на фенотип структурные перестройки делят на сбалансированные и несбалансированные. При сбалансированных перестройках нет избытка или недостатка хромосомного материала, они не приводят к развитию заболевания у носителя. Несбалансированные мутации характеризуются потерей или добавлением генетической информации, и являются причиной заболевания. Сбалансированные структурные перестройки: ● инверсии (inv) – поворот участка хромосомы в ее же пределах на 180 градусов, различают парацентрические (внутриплечевые, при этом меняется рисунок сегментизации) и перицентрические (межплечевые, с участием центромеры), они создают затруднения процесса конъюгации гомологичных хромосом в мейозе, в результате чего у гетерозиготных носителей инверсий происходят частые нарушения образования половых клеток; ● транслокации (t) – перестройка двух хромосом с переносом участка одной хромосомы на другую. Транслокации могут быть простыми, возникающими в результате двух разрывов на двух разных хромосомах, и комплексными, когда в них участвуют три и более хромосом с числом разрывов 4 и более. Различают робертсоновские транслокации (rob), которые образуются при слиянии двух центромер акроцентрических хромосом – центрическое слияние, в результате возникает одна мета- или субметацентрическая хромосома и число хромосом в клетке уменьшается на одну; и реципрокные транслокации (t) – происходит взаимный обмен двух негомологичных хромосом; ● инсерции (ins) – вставка интерстициального фрагмента одной хромосомы в точку разрыва, расположенную в другой хромосоме. По механизму она схожа с транслокацией. Происходит при наличии трех хромосомных разрывов: один разрыв выступает в качестве принимающего участка, а два других – в роли участка-донора. Несбалансированные структурные перестройки: ● дупликации (dup) – удвоение участка какой-либо хромосомы, если удваиваемый участок располагается последовательно, то такая дупликация называется тандемной; ● делеции (del) – утрата части хромосомного материала, может быть концевая или терминальная делеция – потеря дистального участка в результате одного разрыва, может быть интерстициальная делеция – утрата внутреннего сегмента хромосомы при наличии двух разрывов в одном плече; ● нереципрокные (несбалансированные) транслокации – участок хромосомы изменяет свое положение или включается в другую хромосому без взаимного обмена; ● изохромосома (i) – хромосома с двумя идентичными плечами, возникает вследствие аномального деления хромосомы в области центромеры с последующей дупликацией материала короткого или длинного плеча, возникает изохромосома по короткому или длинному плечу. ● дицентрическая хромосома (dic) – возникает вследствие воссоединения двух поврежденных хромосом, несущих центромерные районы, с образованием одной хромосомы с двумя центромерами; ● изодицентрическая хромосома (idic) – изохромосома с двумя близко расположенными центромерными районами; ● кольцевая хромосома (r) – возникает при утрате обоих теломерных участков одной хромосомы с последующим воссоединением открытых концов; ● дериватные или производные хромосомы (der) – хромосомы, возникшие в результате перестроек, затрагивающих две и более хромосомы или в результате множественных перестроек внутри одной хромосомы. Термин «дериват» всегда применяется к хромосоме, имеющей интактную центромеру. 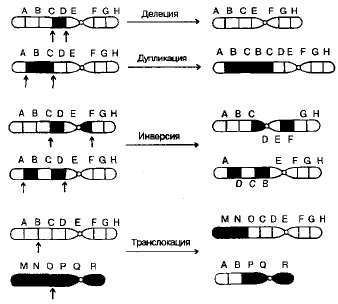 Рис.7. Структурные хромосомные перестройки. 5.3. Геномные мутации Геномные мутации – это числовые хромосомные аномалии, связанные с нарушением численного состава хромосомного набора (кариотипа). К ним относятся полиплоидия и анеуплоидия. Полиплоидия ((2+х) n) - изменение числа хромосом кратное гаплоидному набору, например, триплоидия (3n) и тетраплоидия (4n). В норме в соматических клетках всегда диплоидный набор хромосом – 2n (46) и в половых клетках – гаплоидный (n). У человека зародыши с полиплоидией погибают в ранние месяцы внутриутробного развития. Описаны единичные случаи рождения детей-триплоидов с тяжелыми множественными врожденными пороками развития, несовместимыми с нормальной жизнедеятельностью. Триплоидия может возникать вследствии дигении (оплодотворение диплоидной яйцеклетки гаплоидным сперматозоидом), диандрии (обратный вариант) и диспермии (оплодотворение гаплоидной яйцеклетки двумя сперматозоидами). Анеуплоидия (2n±x) – изменение числа отдельных хромосом в наборе либо в сторону уменьшения (гипоплоидия – вместо двух гомологичных хромосом присутствует одна, это моносомия по данной хромосоме – 2n – 1), либо в сторону увеличения (гиперплоидия – вместо двух гомологичных хромосом имеются их дополнительные копии, трисомия - 2n + 1 или полисомия – 2n + (> 1)). В основе геномных мутаций, как правило, лежит нерасхождение или анафазное отставание хромосом в мейозе. В первом случае нарушается распределение хромосом по дочерним клеткам. Хромосомы, которые в норме должны разделиться остаются соединенными и вместе отходят к одному полюсу. Возможно вторичное нерасхождение – до этого в митозе, простое (однократное) – у 1-го родителя, двойное – у обоих родителей, последовательное – во всех делениях мейоза. Если нерасхождение происходит в двух и более последовательных делениях, то возникают тетра- и пентасомии. Хромосомный мозаицизм (2n ± x/2n) – явление одновременного наличия в организме нескольких клеточных клонов с разным кариотипом. Мозаицизм может возникнуть на любой стадии эмбрионального развития либо в результате митотического нерасхождения хромосом, либо вследствие утраты хромосомы вследствие анафазного отставания. Мозаики с небольшим клоном аберрантных клеток могут иметь не выраженные фенотипические отклонения. 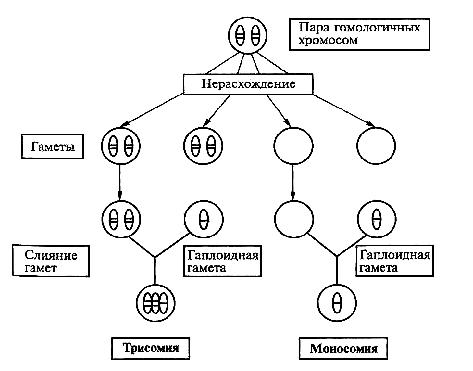 Рис.8. Схема нерасхождения хромосом в мейозе. Контрольно-обучающие вопросы 1. Перечислите виды изменчивости наследственного материала. 2. Какие мутации выделяют в зависимости от их влияния на организм? 3. Что относится к мутациям со сдвигом рамки считывания? 4. Перечислите возможные последствия изменений нуклеотидной последовательности ДНК. 5. Как подразделяются генные мутации в зависимости от их патогенетического эффекта? 6. Какие мутации относятся к сбалансированным структурным перестройкам? 7. Что такое транслокация? 8. Какие виды хромосомных аномалий не встречаются у живорожденных? 9. Опишите механизм возникновения анеуплоидий. 10. .Дайте определение термина «хромосомный мозаицизм». Хромосомные болезни Хромосомные болезни - большая группа клинически многообразных состояний, характеризующихся МВПР, этиология которых связана с количественными или структурными изменениями кариотипа. В настоящее время у человека известно более 700 заболеваний, вызванных изменением числа или структуры хромосом. Около 25 % синдромов приходится на аутосомные трисомии, 35 % - патология половых хромосом и 40 % - структурные перестройки (наиболее часто встречаются транслокации и делеции). Хромосомные болезни составляют 1% педиатрической патологии и являются причиной смертности в детском возрасте в 2,5% случаев. Все хромосомные болезни подразделяют на две большие группы: аномалии аутосом (аутосомные синдромы) и аномалии половых хромосом (гоносомные синдромы). В зависимости от типа мутации классифицируют на синдромы, связанные с числовыми или структурными аномалиями в системе аутосом или половых хромосом. В зависимости от типа клеток, в которых возникла мутация выделяют гаметические мутации, приводящие к полным формам хромосомных болезней, у таких индивидов все клетки несут унаследованную с гаметой хромосомную аномалию; и соматические мутации, возникшие в зиготе или на ранних стадиях дробления, при этом развивается организм с клетками разной хромосомной конституции (два типа и более). Такие формы хромосомных болезней называются мозаичными. Мутация может возникнуть заново в гаметах здоровых родителей (мутация de novo, спорадические случаи). Реже родители уже имели такую аномалию (наследуемые или семейные формы). Большая часть наследуемых случаев хромосомных болезней связана с наличием у здоровых родителей робертсоновских транслокаций, сбалансированных реципрокных транслокаций и инверсий. В этих случаях хромосомные аномалии возникают в связи со сложными перестройками хромосом в процессе мейоза (конъюгация, кроссинговер). Например, у индивидов с синдромом Дауна и трипло-Х образуются гаметы двух типов - нормальные и дисомные. Такое происхождение дисомных гамет - следствие вторичного нерасхождения, т.е. нерасхождения хромосом у индивида с трисомией. Общая характеристика хромосомных болезней: ● для хромосомных болезней типично вовлечение более чем одного гена; ● патология прослеживается, как правило, в пределах одного поколения (преимущественно мутации de novo); ● не следуют менделевским законам наследования; ● множественность поражения: черепно-лицевые дизморфии, врожденные пороки развития внутренних органов и частей тела, задержка физического и психического развития; ● общность клинической картины при разных хромосомных наблюдениях. Фенотипические проявления хромосомных мутаций зависят от следующих главных факторов: ● особенностей вовлеченной в аномалию хромосомы (специфический набор генов); ● типа аномалии (трисомия, моносомия, полная, частичная); ● размера недостающего (при частичной моносомии) или избыточного (при частичной трисомии) генетического материала; ● степени мозаичности организма по аберрантным клеткам; ● генотипа организма; ● условий среды. 8.1. Патогенез хромосомных болезней Несмотря на хорошую изученность клиники и цитогенетики хромосомных болезней, ключевое звено в их развитии еще не выявлено. В основе данной патологии лежит несбалансированность генотипа и нарушение общего генного баланса. Систематизация данных о механизмах нарушений позволило выделить ряд генетических эффектов: ● специфические генетические эффекты - связаны с изменением числа структурных генов, «эффект дозы гена» (при с.Дауна ↑активности супероксиддисмутазы); ● полуспецифические генетические эффекты - изменение числа повторяющихся генов (гены рРНК, тРНК, гистонов, рибосомных белков) - в норме они контролируют ключевые этапы метаболизма клетки, процессов деления, межклеточных взаимодействий; ● неспецифические генетические эффекты - связаны с изменением содержания гетерохроматина в клетке (играет важную роль в клеточных делениях и клеточном росте). Для хромосомных болезней характерен выраженный клинический полиморфизм – от летального эффекта до незначительных отклонений в развитии. 8.2. Аутосомные синдромы Частота аутосомных синдромов составляет 0,7% в популяции, на долю спонтанных абортов в I триместре приходистя 50%, во II-ом триместре - 30%, мертворождений - 5% случаев аномалий аутосом. Несбалансированные мутации в системе аутосом всегда приводят к тяжелым нарушениям в организме человека. Их делят на сублетальные и летальные мутации, они зависят от морфогенетической значимости хромосом. Сублетальные мутации приводят к элиминации большинства эмбрионов в I, II триместре беременности, меньшая часть – к мертворожденнию, еще реже к появлению живорожденных детей с тяжелыми расстройствами органов и систем. К данной группе относятся все частичные моносомии и трисомии аутосом, полные трисомии – 21, 13, 18, 8, 9, 22. Для них характерна клиническая триада: - множественные врожденные пороки развития, - множественные малые аномалии развития, - грубая задержка психоречевого и статико-моторного развития. У больных с большинством аутосомных синдромов отмечается низкая продолжительность жизни. Типична ранняя диагностика болезней. Для летальных мутаций характерна элиминация эмбрионов в I триместре беременности, к ним относятся остальные аутосомы. Синдром Дауна (трисомия хромосомы 21) Первое клиническое описание относится к 1866 году и принадлежит английскому врачу Лангдону Дауну, это наиболее изученная хромосомная болезнь. Частота - 1:600-1:800 новорожденных. Причиной заболевания является наличие добавочного материала 21 хромосомы (критический регион 21q22). Существуют три цитогенетические формы синдрома: ● 93% - регулярная трисомия (47,ХХ (ХУ), +21); ● 2-3% - мозаицизм (47, ХХ,+21/ 46,ХХ); ● 4-5% - транслокации – 15, 14, 21, 22, У, например 46,Х,der (14;21)(q10;q10); 46, ХХ, der (21;22)(q10;q10). Клинический диагноз заболевания прост, и довольно часто ставится новорожденным в родильных домах. Больные с синдромом Дауна имеют характерный фенотип. К основным признакам относятся: низкий рост, брахицефалия, уплощенный затылок, плоский профиль лица, круглое лицо, деформированные ушные раковины, плоская спинка носа, монголоидный разрез глазных щелей, эпикант, макроглоссия, узкое и короткое небо, мелкие зубы, мезобрахидактилия, клинодактилия мизинцев, пятна Брушфильда на радужной оболочке, помутнение хрусталика, сандалевидный промежуток на стопах, часто страбизм, нистагм, выраженная диффузная мышечная гипотония. Изменение дерматоглифики: поперечная («обезьянья») складка на ладони, высокое положение трирадиуса, две кожные складки на мизинце вместо трех. Для больных характерен вторичный иммунодефицит, олигофрения разной степени выраженности. Среди врожденных пороков внутренних органов чаще встречаются пороки сердечно-сосудистой системы (ДМПП, ДМЖП, тетрада Фалло), пороки ЖКТ (атрезия ануса, двенадцатиперстной кишки и др.), мочевой системы (гипоплазия или дисплазия почек, гидроуретер, гидронефроз). В связи с измененным иммунитетом у больных частые респираторные инфекции, часто лейкозы. Лечебная помощь детям с синдромом Дауна неспецифична. Постоянно проводится общеукрепляющее лечение. Должно быть полноценное питание. Врожденные пороки сердца устраняют оперативно. Если нет тяжелых пороков развития внутренних органов, то продолжительность жизни значительно больше, чем при других аутосомных трисомиях (до 20 – 30 лет и более). В настоящее время успешно разрабатываются принципы коррекции таких детей, позволяющие социально адаптировать их к жизни. Синдром Эдвардса (трисомия хромосомы 18) В 1960 г. Эдвардс с сотрудниками при цитогенетическом исследовании клеток больных с множественными пороками развития обнаружили лишнюю хромосому из группы Е. В дальнейшем эту хромосому определили как 18, и трисомию стали называть синдромом Эдвардса. Почти во всех случаях синдром Эдвардса обусловлен простой трисомией (гаметическая мутация у одного из родителей), реже встречаются мозаичные формы (нерасхождение на ранних стадиях дробления), транслокационные формы крайне редки. Частота - 1:5000- 1:10000 новорожденных. Девочки страдают данным синдромом значительно чаще, чем мальчики (М1:ЖЗ). К характерным признакам относятся: пренатальная гипоплазия (масса при рождении не более 2200г), «птичье лицо», «стопа-качалка», долихоцефалия, флексорное положение кистей, нижняя челюсть и отверстие рта маленькие, глазные щели узкие и короткие, деформированные и низко расположенные ушные раковины, тонкая переносица, эпикант, короткая шея и грудина, синдактилия. Среди пороков внутренних органов чаще всего встречаются пороки сердца и крупных сосудов (ДМЖП, ДМПП), пороки ЦНС (спинномозговая грыжа, гипоплазия мозолистого тела, гипоплазия мозжечка), пороки ЖКТ (дивертикул Меккеля, атрезия пищевода), пороки мочевой системы (удвоение почек и мочеточников, кисты, гидро- и мегалоуретер) и др. Нарушение развития головного мозга наблюдают во всех случаях. У детей отмечается тяжелая задержка психомоторного и физического развития, затруднения при глотании, проблемы с кормлением. Большинство больных погибают до 1 года. Синдром Патау (трисомия хромосомы 13) Синдром впервые был описан в 1960 году Патау и с тех пор носит название автора. Частота - 1:7000 новорожденных. Выделяют три цитогенетические формы: простую трисомию – 47,ХХ (ХУ),+13 (75%), транслокационные варианты (20%), чаще – 46, ХХ (ХУ),der(13;13)(q10;q10) и мозаики (5%). Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией, беременность у женщин осложнена многоводием. Фенотипически выделяют триаду признаков: микрофтальмия (реже анофтальмия), расщелина губы и неба, полидактилия (чаще гекскадактилия). Из других внешних признаков отмечаются микроцефалия, тригоноцефалия, широкий плоский нос, флексорное сгибание кистей, дефекты скальпа. При синдроме Патау центральная нервная система поражена во всех случаях: аринэнцефалия, аплазия мозолистого тела, голопрозэнцефалия, гипоплазия червя мозжечка. Встречаются пороки костной, сердечно-сосудистой системы (ДМПП, ДМЖП, коарктация аорты), поликистоз почек, гидронефроз, незавершенный поворот кишечника, омфалоцеле, крипторхизм и др. В ряде случаев наблюдают грубые пороки лица – циклопия, пробошизис, цебоцефалия, связанные с голопрозэнцефалией. У детей отмечается глубокая задержка умственного и физического развития. Продолжительность жизни больных резко снижена, обычно дети погибают в первые дни или недели жизни. В настоящее время клинически описаны случаи частичной трисомии и частичной моносомии для каждого плеча всех аутосом (кроме коротких плечей акроцентрических хромосом), это так называемые синдромы, обусловленные перестройками аутосом. При кариотипировании выявляются делеции или дупликации соответствующих плечей хромосом. Фенотип определяется потерей или удвоением «критического сегмента». Данные нарушения могут быть результатом мутации de novo, сегрегации перестроенных хромосом в мейозе у родителей-носителей сбалансированных хромосомных перестроек (инверсии, реципрокные транслокации). Синдром «крика кошки» (моносомия короткого плеча хромосомы 5) Впервые клинические проявления синдрома описал Лежен в 1963 году. Причиной заболевания является частичная моносомия по короткому плечу 5 хромосомы. Кариотип: 46, ХХ (ХУ),dе1(5р). Реже встречаются кольцевые хромосомы – r(5), мозаицизм. Ответственен за развитие синдрома сегмент 5р15.1- р15.2. Частота - 1:45000-50000 новорожденных. Дети, как правило, рождаются после нормально протекавшей беременности. В неонатальном периоде состояние резко ухудшается: приступы цианоза, инспираторный стридор, снижение двигательной активности, угнетение сосательного рефлекса, рвота. Наиболее характерным является специфический крик новорожденного, напоминающий кошачье мяуканье, отсюда и название синдрома. Это связано с недоразвитием гортани, надгортанника, изменением хрящевой ткани, ларингомаляцией. У больных детей отмечается пренатальная гипотрофия, микроцефалия, мышечная гипотония, лунообразное лицо, широкая переносица, антимонголоидный разрез глаз, гипертелоризм, высокое небо, микрогения, эпикант, низко расположенные деформированные ушные раковины, короткий фильтр. Аномалии скелета: клинодактилия V пальцев, синдактилия, плоскостопие, сколиоз. Пороки внутренних органов негрубые, часто наблюдаются врожденные пороки сердца, почек, крипторхизм. Глубокая умственная отсталость. Витальный прогноз относительно благоприятный, описаны случаи продолжительности жизни до 40 лет. Высокий генетический риск синдрома отмечается при носительстве реципрокной перестройки 5 хромосомы у родителей. Синдром Вольфа-Хиршхорна (моносомия короткого плеча хромосомы 4) Причиной синдрома является частичная моносомия по короткому плечу 4 хромосомы (4р-). Варианты кариотипа: 80 % - делеция, остальные случаи – t, r. Критический сегмент – 4р16. Популяционная частота 1:100000. Для этого синдрома характерны микроцефалия, гипертелоризм, широкое межбровное пространство, эпикант, экзофтальм, выступающие надбровные дуги, крупный нос, иногда клювовидный. Лицо в виде «маски греческого воина», рот с опущенными углами, короткий фильтр. Ушные раковины крупные, оттопыренные, низко расположенные. Шея короткая тонкая. Пальцы длинные, с заостренными концевыми фалангами, ногти выпуклые. Из пороков внутренних органов чаще отмечаются пороки сердца, почек, ЖКТ. Характерным признаком синдрома является наличие неправильной формы углубления в области крестца (sinus sacralis). У детей наблюдается резкая задержка психомоторного развития, частые судороги. 8.3. Синдромы, обусловленные аномалиями в системе половых хромосом (гоносомные синдромы) Общие особенности синдромов, связанных с аномалиями половых хромосом: ● встречаются полные моносомии (45,Х), трисомии (Х, У), полисомии (Х, У); ● страдают гонады; ● отсутствие множественных ВПР и МАР; ● продолжительность жизни не снижена; ● интеллект нормальный или незначительно снижен, или изменен качественно; ● протекают легче по сравнению с аномалиями аутосом; ● диагностика в более поздние сроки (пубертатный период); ● клиническая картина включает нарушение роста, полового развития и репродуктивной функции; ● прогноз для жизни благоприятный. Синдром Шерешевского-Тернера (моносомни Х-хромосомы) Данный синдром характеризуется полной или частичной моносомией Х. Впервые описал Н.А.Шерешевский в 1925году, в 1930 г. – О.А.Ульрих, 1938 году Х.Тернер выделил основную триаду признаков. Популяционная частота синдрома составляет в среднем 1:5000 новорожденных девочек. Данный синдром является наиболее тяжелым среди болезней, обусловленных аномалиями в системе половых хромосом. Цитогенетические варианты: - 45, Х - классический вариант - 57%; - мозаики 45,Х/46,ХХ; 45,Х/47,ХХХ – 12%; - мозаики 45,Х/46,ХY – 4%; - 46,X,i(Xq) и мозаики с линиями клеток i(Xq) – 17%; - 46,X,del(Xq) и мозаики с линиями клеток del(Xq) - 1%; - прочее [dеl (Хр), г(Х), мозаики] – 9%. В отличие от других гоносомных синдромов при синдроме Шерешевского-Тернера в раннем возрасте отмечается значительное отставание в росте, сочетающееся с многочисленными микроаномалиями: антимонголоидный разрез глаз, эпикант, миопия, низко расположенные ушные раковины, высокое небо, неправильный рост зубов и прикус, низкий уровень роста волос на шее, укорочение IV пальцев на кистях, вальгусная деформация локтевых суставов, своеобразная форма ногтей, широкая грудная клетка, гипертелоризм сосков, обилие пигментных пятен, Х-образная деформация голеней. В первые месяцы среди ранних симптомов заболевания встречаются лимфатические отеки тыльных поверхностей кистей и стоп, крыловидные кожные складки на шее, «лицо сфинкса». Пренатально по УЗИ выявляют маркер – гигрому шейной области. В пубертатный период отмечается выраженный половой инфантилизм: отсутствие молочных желез, скудное вторичное оволосение, первичная аменорея. Эти явления связаны с дисгенезией гонад, с отсутствием яичников, что ведет к бесплодию. У части больных отмечаются негрубые пороки внутренних органов: пороки сердца, коарктация аорты, пороки почек. Конечный рост женщин с данным синдромом составляет 135-141 см. Интеллект не снижен. Характерно отсутствие полового хроматина при классическом варианте кариотипа. Синдром Клайнфельтера (дисомия Х при мужском кариотипе) Впервые синдром описан в 1942 г. Клайнфельтером. Характеризуется добавочными Х-хромосомами в мужском кариотипе. Частота – 1: 700 - 1000 новорожденных мальчиков. Выделяют несколько вариантов кариотипа: ● трисомный вариант - 47, ХХУ: нерасхождение Х-хромосомы в мейозе; ● полисомные варианты - 48,ХХХУ: а) последовательное нерасхождение самих хромосом, затем хроматид, б) сочетанное нерасхождение у обоих родителей; - 48,ХХУУ – последовательное нерасхождение в одном делении – Х, в другом У-хромосомы; - 49, ХХХХУ, 49,ХХХУУ – сочетанное и последовательное нерасхождение; ● редко мозаичные варианты. У больных с трисомным вариантом кариотипа характерна клиническая триада: гипогонадизм, гинекомастия, бесплодие. Для больных с данной цитогенетической формой характерна более мягкая клиническая картина. Вследствие гормонального дисбаланса такие больные имеют высокий рост, евнухоидные пропорции (узкие плечи, широкий таз), скудное оволосение. Уровень гонадотропинов в моче соответствует норме или повышен. Снижен уровень тестостерона. В дизгенетических яичках семенные канальцы замещены соединительной тканью и гиалинизированы. У всех взрослых больных азоспермия, олигоспермия. Диагноз синдрома чаще выставляется в пубертатный период или в постпубертатном возрасте. Больные имеют психологические и характерологические особенности, такие как внушаемость, слабоволие, вспышки агрессивности, инфантильность, склонность к немотивированным поступкам и к алкоголизму, лабильность характера, эмоциональные срывы, аутизм, гипосоциальность. При полисомиях клиническая картина более тяжелая. Пол мужской, значимо задержан. Отмечается выраженная олигофрения, микроорхидизм, микрогенитализм, крипторхизм, выраженная задержка полового развития, качественное обеднение интеллекта, олигофрения, бесплодие, множественные микроаномалии, что делает таких больных похожими на больных с аутосомной патологией. При синдроме Клайнфельтера выявляется половой хроматин. Синдром трисомии Х (Triple-Xsyndrome) Наличие трех Х-хромосом при отсутствии У-хромосом проявляется синдромом Х. Частота – 1: 1000 новорожденных девочек. Является наиболее сложным синдромом для диагностики из всех болезней, связанных с аномалиями в системе половых хромосом. Варианты кариотипа: ● 47, ХХХ, реже мозаицизм; ● полисомии Х: 48, ХХХХ, 49, ХХХХХ. Самым частым симптомом трисомии Х является умственная отсталость в стадии дебильности. С возрастом увеличивается в два раза риск заболевания каким-либо психозом. Часто у взрослых женщин наблюдается шизофрения с неблагоприятным течением, с выраженными изменениями личности. Возможно развитие эпилепсии как в детском, так и в более старшем возрасте. Соматические аномалии выражены слабо. Иногда можно наблюдать микроцефалию, эпикант, гипертелоризм, косоглазие, высокое небо, уплощение переносицы, кифосколиоз. Возможно нарушение менструального цикла, снижение фертильности. При отсутствии олигофрении диагноз трисомии X может быть не поставлен в течение всей жизни, или она может быть обнаружена случайно. При полисомиях клиническая картина значительно более тяжелая. Отмечается выраженная олигофрения, эпилепсия, множественные микроаномалии: эпикант, гипертелоризм, клинодактилия, короткий фильтр, высокорослость, нарушение полового развития (аменорея), бесплодие, гипокинезия, врожденные пороки сердечно-сосудистой системы (незаращение артериального протока). Синдром дисомии У (синдром дубль - У) Частота 1- 1,5 : 1000 новорожденных мальчиков. Варианты кариотипа: ● 47,ХУУ, реже мозаичные формы; ● полисомии У: 48, ХУУУ, 49, ХУУУУ. Для выявления добавочной У-хромосомы используется люминесцентное окрашивание. Наиболее характерными симптомами синдрома являются высокий рост (186 см и выше), макроцефалия, увеличение конечностей, длинные веретенообразные пальцы, грубые черты лица (по типу акромегалии), плотное телосложение, нарушение половой дифференцировки (крипторхизм, гипогонадизм, дисплазия гениталий), нарушение поведения (агрессивность, антиобщественные поступки, психопатические черты характера, импульсивность). Чаще половая функция не страдает. У больных с полисомией У отмечается умственная отсталость, множественные малые аномалии развития, пороки крупных сосудов (стеноз легочной артерии), неврологические, психические расстройства. 8.4. Синдромы микроструктурных перестроек (микроцитогенетические синдромы) Данные синдромы связаны с микроструктурными перестройками хромосом, размер которых не превышает 3-5 млн п.н., что часто делает их невидимыми при использовании светового микроскопирования. Они также получили название синдромы «протяженного гена» - захватывают несколько генов (микроделеции, микродупликации строго определенных участков хромосом). Незначительные нарушения в структуре хромосом могут быть выявлены с помощью молекулярно-цитогенетических и молекулярных методов. Эти синдромы клинически были описаны задолго до того, как была выявлена их хромосомная этиология. По клинической картине и наследованию они схожи с генными заболеваниями. В настоящее время выяснена этиология около 20 нозологических форм, при которых выявлены микроструктурные хромосомные нарушения. Их частота в среднем составляет 1: 50000 – 100000 новорожденных. Показано, что микроструктурные аномалии хромосом сопровождают не только синдромы МВПР, но и различные гиперпластические процессы (злокачественные образования, например, ретинобластома). Каждый синдром имеет четкую и специфическую клиническую картину. К синдромам этого класса относятся: с.Ангельмана – микроделеция 15q11.2-q13 (в хромосоме от матери), с. трихо-рино-фалангеальный 2-го типа (Лангера-Гидиона) – микроделеция 8q24.11-q24.13, с. Прадера-Вилли - 15q11-q12 (в хромосоме от отца), с. Беквита-Видемана микродупликация 11p15.5. Контрольно-обучающие вопросы 1. На какие группы подразделяют все хромосомные болезни? 2. Дайте общую характеристику хромосомных болезней. 3. От каких факторов зависят фенотипические проявления хромосомных мутаций? 4. Перечислите возможные исходы аномалий в системе аутосом. 5. Укажите возможные формулы кариотипа при синдроме Дауна. 6. Перечислите клинические особенности при синдроме Патау. 7. Назовите «критический» сегмент, мутации в котором обуславливают клинические проявления синдрома «крика кошки». 8. Опишите общие особенности синдромов, связанных с аномалиями половых хромосом. 9. Укажите возможные формулы кариотипа при синдроме Шерешевского-Тернера. 10. С помощью, каких методов исследований возможна диагностика микроцитогенетических синдромов? |