реф - АКРИЛОНИТРИЛ. Содержание Введение 2 Основные свойства 3 displaystyle mathsf ch 2textCHtextch 3nh 31,5O 2rightarrow ch 2textCHtextCN3H
 Скачать 1.25 Mb. Скачать 1.25 Mb.
|

|
Содержание Введение 2 1. Основные свойства 3 {\displaystyle {\mathsf {CH_{2}{\text{=}}CH{\text{-}}CH_{3}+NH_{3}+1,5O_{2}\rightarrow CH_{2}{\text{=}}CH{\text{-}}CN+3H_{2}O}}}2. Анализ методов синтеза акрилонитрила 4 Заключение 18 Список использованной литературы 19 ВведениеОдин из важнейших и значимых классов органических соединений - нитрилы занимает важное место как в области теоретических исследований в органической химии и нефтехимии, так и в прикладном плане. Тем не менее анализ литературных данных последних 30-ти лет показывает, что имеется только две-три фундаментальные работы или претендующие на фундаментальность попытки обобщить существующий материал по этому вопросу. Среди них необходимо упомянуть работы, которые, однако, уже более относятся к истории вопроса зарождения и развития химии нитрилов. Поэтому рассмотрение прогресса в этом направлении органической химии, систематизация вновь появившихся данных было бы полезным и своевременным в настоящий момент. В связи с этим анализ методов получения нитрилов и реакций на их основе, на наш взгляд, является важным и актуальным для формирования рекомендаций на перспективу развития химии и технологии нитрилов. Настоящая работа посвящена анализу методов получения акрилонитрила как наиболее известного и часто используемого представителя класса нитрилов. Литературные данные содержат информацию о ряде основных методов получения акрилонитрила. 1. Основные свойстваАкрилонитрил (цианистый винил, винилоцианид, проп-2-енонитрил), CH2=CH-C≡N — нитрил акриловой кислоты. Применяется при производстве некоторых видов синтетического каучука. Путём полимеризации акрилонитрила в полиакрилонитрил и последующего прядения получают синтетические волокна, например нитрон, или модакриловые волокна. Бесцветная жидкость с характерным запахом миндаля или вишневых косточек, растворима в воде, т. кип. 77 °C. Пары тяжелее воздуха. Относится к категории СДЯВ (сильнодействующих ядовитых веществ). Вещество, способное вызывать аллергические заболевания в производственных условиях. ПДК в воздухе рабочей зоны: ПДК м.р.= 1 мг/м3, ПДК с.с.= 0,5мг/м3. Канцерогенное вещество. Полиакрилонитрильные волокна и нити (ПАН) в настоящее время представляют наиболее распространенный вид промышленно освоенных карбоцепных синтетических волокон. Это связано с особыми свойствами ПАН-волокна: низким коэффициентом теплопроводности, пушистостью, объемностью, которые делают ПАН-волокна практически равноценными заменителями шерсти. Кроме того, этот полимер при определенных условиях обладает способностью к циклизации, что определяет такой ассортимент производства ПАН, как технический жгутик, используемый в качестве сырья для углеродных волокон. Термин «полиакрилонитрильное волокно» обычно употребляется по отношению к волокнам, содержащим не менее 85% звеньев акрилонитрила (АН). По отношению к волокнам, доля АН в которых составляет от 35% до 85%, употребляется термин «модакриловое (modacrylic) волокно». Модакриловые волокна содержат значительное количество галогенпроизводных мономеров и применяются в случаях, когда необходимо повысить огнестойкость материала. Технологический процесс получения ПАН включает следующие основные стадии: синтез полиакрилонитрила, получение прядильного раствора и подготовка его к формованию - это осуществляется в химическом цехе производства; формование, ориентационное вытягивание и отделка волокна или нити - прядильно-отделочный цех; регенерация растворителя - цех регенерации растворителя. {\displaystyle {\mathsf {CH_{2}{\text{=}}CH{\text{-}}CH_{3}+NH_{3}+1,5O_{2}\rightarrow CH_{2}{\text{=}}CH{\text{-}}CN+3H_{2}O}}}2. Анализ методов синтеза акрилонитрилаТак, в 1983 году Муре получил это вещество путем отщепления воды от этиленциангидрина, а также дегидратацией амида акриловой кислоты в присутствии фосфорного ангидрида: HO(CH2)2CN⎯⎯→CH2 = CH −CN +HOH CH2 = CH − CONH2 ⎯⎯→CH2 = CH − CN + HOH В работе с выходом 67% был получен акрилонитрил каталитической дегидратацией акриламида при пропускании паров последнего над нагретым до 450-500°С пиролюзитом. По аналогии с упомянутой работой Кунга (1945 г.) в работе отщепляли в одну стадию воду от амида β-оксипропионовой кислоты, который, в свою очередь, можно получить с выходом 50% из соответствующего лактона при взаимодействии последнего с избытком жидкого аммиака: CH −CH −C=O+ NH3 ⎯⎯→ ⎯⎯→CH2(OH)−CH2 −CONH2 Al2⎯O⎯3 CH2(OH) − CH2 − CONH2 ⎯⎯ → Al2⎯O⎯3 CH2 = CHCN + 2H2O ⎯⎯ → Аналогичная реакция возможна при отщеплении молекулы спирта от амида β-алкоксипропионовой кислоты или β-алкоксиизомасляной кислоты в присутствии двуокиси кремния с добавками небольшого количества окиси тантала, циркония или вольфрама: C2H5O−CH2 −CH2 −CO−NH2 ⎯450⎯ →⎯0C 4500C ⎯⎯ →⎯ CH2 =CH−CN+C2H5OH+H2O Также возможна дегидратация альдоксимов. Водоотнимающим агентом чаще всего является уксусный ангидрид. Этим методом акрилонитрил получается количественно из акролеиноксима. Последний получается пропусканием оксима пропионового альдегида над катализатором (смесь боксит – окись хрома) при температуре 400-500°С. Помимо методов дегидратации, рядом авторов часто применялся и метод дегидрирования пропионитрила, в результате которого также образовывался акрилонитрил при температуре реакции 600-625°С и участии окислов хрома или ванадия в качестве катализаторов: CH3 − CH2 − CN ⎯⎯→CH2 = CH − CN + H2 Реакция протекает с более высоким выходом, если она проводится при более высокой температуре, а в качестве катализаторов дегидрирования используются сульфиды вольфрама и никеля [16] либо молибденовые катализаторы [17]. Помимо пропионитрила в реакцию дегидрирования может вступать и аллиламин, что также приводит к образованию акрилонитрила [18]: CH2 = CH −CH2 − NH2 ⎯⎯→CH = CH −CN + 2H2 Вовлечение пропионитрила в реакцию окисления воздухом в газовой фазе в присутствии катализатора тоже приводит к акрилонитрилу. Катализаторами реакции являются тонкоизмельченная медь или серебро, при этом протекание реакции облегчается добавлением 2% йода [19] или бромистого водорода [20], степень превращения 25%, выход акрилонитрила 80-85%. Температура процесса 600-610°С: CH3 −CH2 −CN+1/2O2 ⎯⎯→CH2 =CH−CN+H2O В реакции каталитического окисления аллиламина [21] в присутствии водяных паров при температуре 450-600°С над серебром, нанесенным на карбид кремния, помимо акрилонитрила (с выходом 90%) образуются аммиак, углекислый газ, ацетон и синильная кислота: CH2 = CH − CH2 − NH2 +1/ 2O2 ⎯⎯→ ⎯⎯→H2O + CH2 = CH − CN Одним из методов получения непредельных нитрилов является дегидрохлорирование хлоросодержащих насыщенных нитрилов в жидкой фазе в присутствии третичных оснований (пиридин) [22,23] при температуре 150-250°С. Однако, отщепление хлористого водорода лучше осуществлялось в газовой фазе в присутствии катализаторов [24]. Температура процесса колебалась от 575 до 625°С. При этом процессы протекали с различными выходами в зависимости от используемых катализаторов – двуокиси титана [25-28], хлорного железа [29] или гидрохлорида органического основания [30]: CH3 − CHCl − CN ⎯⎯→CH2 = CHCN + HCl Если реакцию вести в присутствии метилового спирта, то вместо хлористого водорода [30] образуется хлористый метил. Кроме того, в 40-х и 50-х годах прошлого столетия были разработаны методы получения акрилонитрила разложением эфиров этиленциангидрина и ароматических сульфоновых кислот. Метиловый эфир этиленциангидрина, нагретый до 300-350°С в стеклянной или фарфоровой трубке дает до 80% акрилонитрила и метилового спирта, а этиловый эфир этиленциангидрина в контакте с безводной ортофосфорной кислотой, нанесенной на пемзу при 360-400°С также дает 80-90% выход акрилонитрила и этиловый спирт [31,32]. Киемо и Уэлтон показали [33], что при нагревании со щелочами пара-толуолсульфоната этиленциангидрина также образуется акрилонитрил: CH3C6H4SO2OCH2CH2CN ⎯⎯→ ⎯⎯→CH3C6H4SO3H + CH2 = CH − CN Кроме того, акрилонитрил получали пиролизом нитрила янтарной кислоты при 500-600°С в присутствии инертного газа [34]: (CH2CN)2 ⎯⎯→CH = CH − CN + HCN а также пиролизом β,β′-дициандиэтилового эфира [35,36] при температуре 400-500°С в присутствии солей щелочных металлов слабых кислот (формиата калия, ацетата калия, натрия, фосфата натрия, тетрабората натрия и др.). При этом выход конечного продукта достаточно высокий – 94%: O〈 CHCH22CHCH22CNCN→ 2CH2 = CH − CN + H2O При разложении некоторых аминопроизводных пропионитрила, например, при сухой перегонке соли β-диэтиламинопропионитрила при температуре 160-190°С был получен солянокислый диэтиламин и акрилонитрил (77%) [37], а при разложении при температуре 200 0С β,β′-аминодипропионитрила путем плавления с фталевым ангидридом был также получен акрилонитрил (80%) [38]: 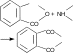 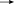 CO CH2CH2CN CO CH2CH2CNCH2CH2CN N - CH3 - CH2 - CN + H2O + CH2 = CH - CN Дучер и Уолк в 1949 г. запатентовали [39,40] способ получения нитрилов пропусканием синильной кислоты и галлоидных алкилов или алкенов через нагретый до 500°С силикагель с последующим охлаждением газов до 100°С. Так, из синильной кислоты и дихлорэтана или хлористого винила получали акрилонитрил: CH2Cl−CH2Cl+HCN⎯⎯→CH2 =CH−CN+2HCl CH = CHCl+ HCN⎯⎯→CH2 = CH −CN + HCl Таким же способом акрилонитрил получали [41] из винилацетата и синильной кислоты с 94% выходом: CH =CHOCOCH3 +HCN⎯⎯→ ⎯⎯→CH2 =CH−CN+CH3COOH В конце 50-х годов ХХ века были предложены другие способы получения акрилонитрила [42-44], одним из которых являлся метод кратковременного нагревания смеси синильной кислоты с этиленом или пропаном до температуры 1000 °С в среде инертного газа, причем соотношение углеводорода и синильной кислоты было 10:1. Было показано, что избыток углеводорода можно рециркулировать в реактор. В этих условиях выход акрилонитрила составлял 78-80%, также получалось небольшое количество ацетонитрила и пропионитрила. Авторами работ [45-49] было показано, что вместо синильной кислоты в этой реакции может быть использован дициан или хлорциан [50]. А для снижения температуры процесса можно применять катализаторы – алюмосиликаты [51,52], а также окись алюминия, обработанную фтористым бором. 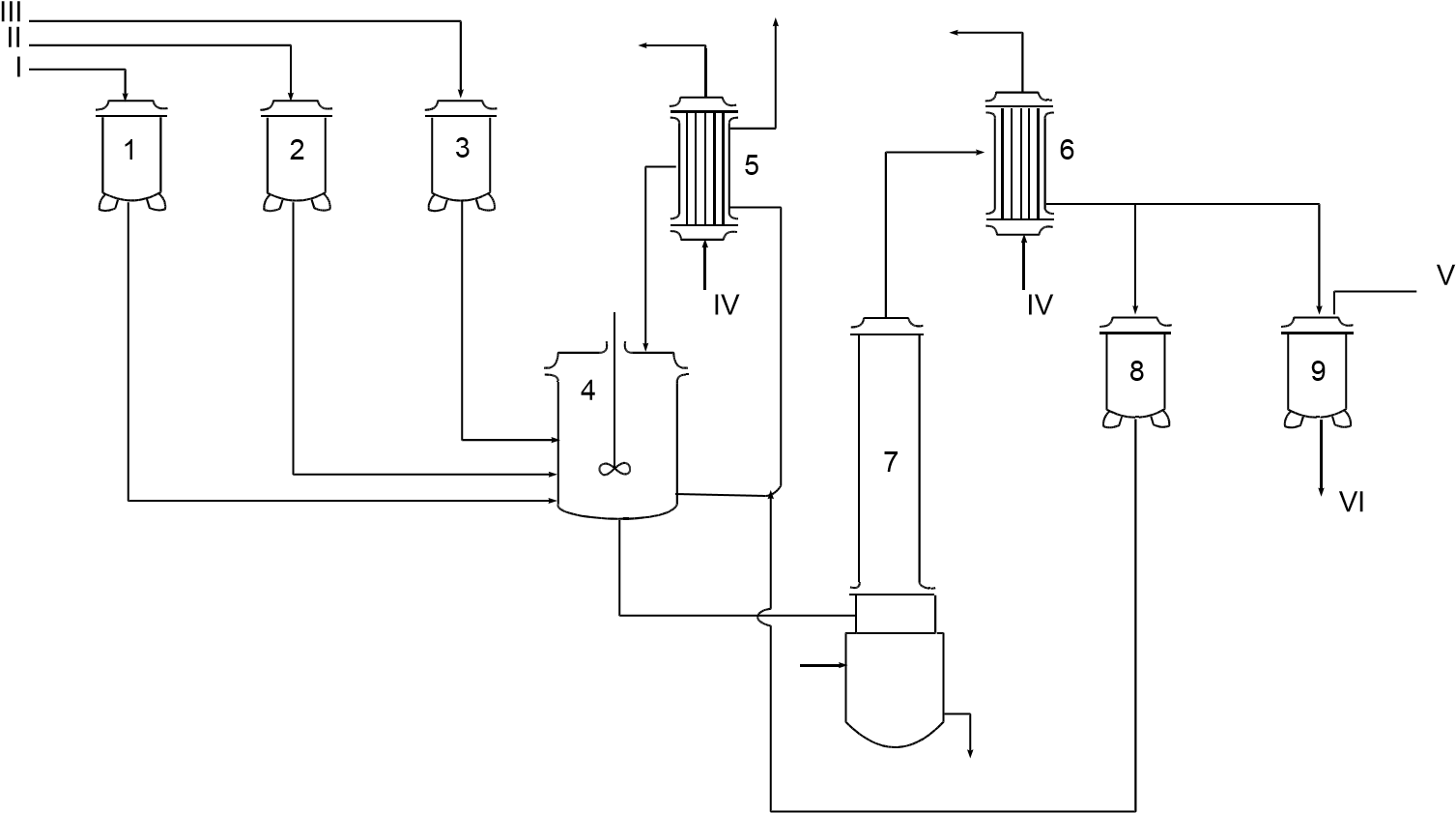 Рис.2 Технологическая схема получения этиленциангидрина. I – окись этилена, II – синильная кислота, III – катализатор, IV – охлаждение, V – вакуум, VI – этиленциангидрин. 1,2,3 – мерники, 4 – реактор, 5,6 – конденсаторы, 7 – отпарная колонна, 8,9 – сборники. Рядом авторов акрилонитрил был получен с выходом 60-80% обработкой кислородом соответствующего альдегида при 30°С в метанольном растворе, содержащем аммиак, сильное основание и каталитически активный комплекс меди или тетраацетат свинца. Привлекает внимание и метод пиролиза изобутиронитрила без катализатора при температуре 650-725°С и времени контакта 4-60 сек с образованием одновременно акрилонитрила и метакрилонитрила. Интересным способом была проведена реакция термической конденсации ацетонитрила и синильной кислоты при 800-1000°С. В результате наряду с фумаронитрилом и малононитрилом образуется и акрилонитрил. Проанализировав перечисленные реакции синтеза непредельных нитрилов, можно было бы предложить классификацию основных методов синтеза в виде схемы (рис.1). 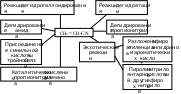 Рис.1. Схема методов получения акрилонитрила. Надо отметить, что это в основном каталитические реакции. Оказалось, что наиболее привлекательными в плане промышленной реализации являются реакции дегидратации, дегидрирования, дегидрохлорирования, окисления и аммонолиза. Если рассмотреть дегидратацию этиленциангидрина, то немаловажное значение имело получение этиленциангидрина, в основном из окиси этилена и синильной кислоты в присутствии катализаторов основного характера (карбонаты и цианиды щелочных и щелочноземельных металлов, амины алифатического и ароматического рядов и аминоспирты. Технологическая схема производства этиленциангидрина приведена на рис.2. Анализ выявил несколько методов получения непредельных нитрилов, в частном случае акрилонитрила. Метод дегидратации является одним из наиболее важных и одновременно перспективных. На рис.3 представлена схема дегидратации этиленциангидрина. Этот способ, предложенный Муре, осуществлялся в присутствии фосфорного ангидрида и позволял получать ацетонитрил с выходом 60%. К катализаторам дегидратации этиленциангидрина можно отнести и хлористый цинк, бисульфит натрия, пемзу со следами окиси олова, металлическое олово, углекислый натрий, сульфат магния, кислый фосфат аммония, окись или гидроокись кальция, стеарат натрия, окись магния, борат натрия, формиат натрия с добавкой водорастворимого противоионного вещества, насыщенный раствор хлористого натрия при пропускании через эту смесь углекислого газа. Надо отметить, что в этих работах было описано 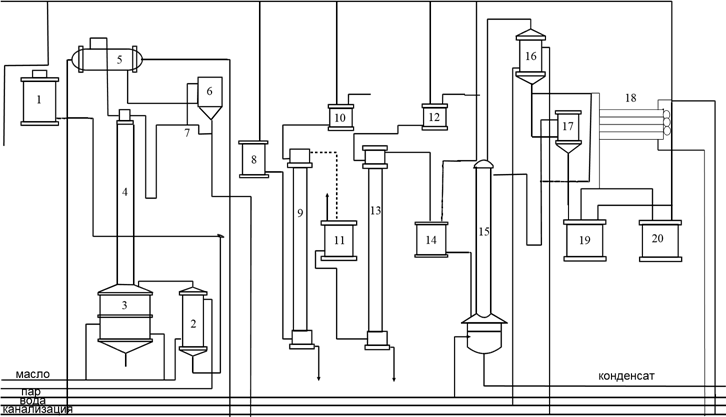 Рис.3. Технологическая схема дегидратации этиленциангидрина: 1 – бак, 2 – теплообменник, 3 – реактор, 4 – колонна, 5 – холодильник, 6 – флорентин, 7 – делитель флегмы, 8 – приемник, 9 – промывная колонна, 10,11,12,14 – флорентины, 13 – колонна, 15 – колонна азеотропной разгонки, 16 – дефлегматор, 17 – флорентин, 18 – холодильник, 19, 20 – емкости. Из-за образования и накопления этих продуктов реакции затруднено непрерывное протекание реакции, а также происходит загрязнение конечных продуктов. Фирма “American Cyanamid” запатентовала способ дегидратации этиленциангидрина в среде инертного жидкого углеводорода с целью растворения накапливающихся в реакторе смол, что позволило проводить непрерывный процесс и повысить выход акрилонитрила до 80-85%. Технологическая схема происпользование образующейся в результате дегидратации реакционной смолы в качестве катализирующей добавки. Оказалось, что использование этой смолы и формиата натрия при температуре 2100С способствует повышению выхода акрилонитрила до 90%. Осуществление процессов дегидратации связано с протеканием и образованием побочных реакций и соединений. При дегидратации этиленциангидрина также может протекать реакция гидролиза акрилонитрила и этиленциангидрина: CH2 = CH −CN + 2H2O⎯⎯→ ⎯⎯→CH2 = CH −COO+ NH3 HO − CH2 − CH2 − CN + 2H2O ⎯⎯→ ⎯⎯→CH2(OH) − CH2 − COOH + NH3 Наличие аммиака способствует протеканию реакции с акрилонитрилом, образуя при этом аминопропионитрил: CH2 = CH−CN+ NH3 ⎯⎯→H2N−CH2 −CH2 −CN А наличие щелочной среды способствует протеканию реакции взаимодействия с этиленциангидрином с образованием β,β′-дицианэтилового эфира [86-88]: CH2(OH) − CH2(CN) + CH2 = CH − CN ⎯⎯→ ⎯⎯→O〈CHCH22CHCH22CNCN А гидролиз этого эфира приводит к образованию высоковязких аминокислот или их солей: NC−CH2 −CH2 −O−CH2 −CH2 −CN+3H2O⎯⎯→ ⎯⎯→NH2 −CO−CH2 −CH2 −O−CH2 −CH2 −CONH2 цесса дегидратации этилен- циангидрина представлена на рис.3. Регенерация катализатора проводится продувкой воздуха при температуре 500°С с частичным возвратом его активности, а частые регенерации способствуют его разрушению. Метод использовался на ряде фирм США, но отсутствие устойчивого активного катализатора, высокая стоимость окиси этилена уменьшают вероятность применения данного способа. Однако в 1960 г. 14% всего акрилонитрила было получено именно из окиси этилена. На рис. 4 представлена схема процесса получения акрилонитрила из окиси этилена. 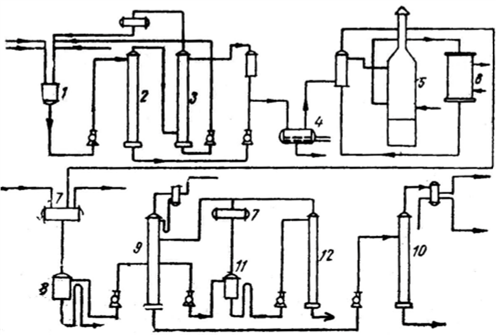 Рис.4. Схема получения акрилонитрила из окиси этилена: 1-этиленциангидриновый реактор; 2 – колонна отгонки легколетучих продуктов; 3 – абсорбер; 4 – испаритель; 5 – перегреватель; 6 – акрилонитрильный реактор; 7 – конденсаторы; 8, 11 – сепараторы; 9 – колонна дегазации; 10 – ректификационная колонна; 12 – отгонная колонна. Поэтапно этот процесс можно представить так: перегонка CH2 −CH2 +HCN→CH2OH−CH2CN⎯⎯⎯⎯⎯⎯→ \ / 85−90% Sn O перегонка ⎯⎯⎯⎯⎯⎯→H2O+CH2 =CH−CN Sn 75−80% Таким этот процесс был принят в производство в Германии. Процесс осуществим как в паровой, так и в жидкой фазе, жидкофазная дегидратация осуществляется в аппарате периодического действия нагреванием в смеси с MgCO3 при температуре 170-250°С или в среде высококипящего органического растворителя. А парофазная дегидратация этиленциангидрина осуществляется прямым пропусканием его паров через реактор непрерывного действия, загруженный катализатором дегидратации. Для упрощения процесса смесь окиси этилена и синильной кислоты пропускали при 250о над катализатором (смесь окиси алюминия и силикагеля), при этом выход акрилонитрила составлял 70-80%. В Великобритании в 40-е годы был разработан метод термического разложения ацетата ацетальдегидциангидрина: OH CH3COOHOCOCH3   CH3 - CH + (CH3CO)2O CH3 - CH + (CH3CO)2O  CH3 - CH CH3 - CHCN CH2 = COCN Ацетат ацетальдегидциангидрина получали и через винилацетат: CH2 = CH − OCOCH3 + HCl ⎯⎯→ Cl | → CH3 − CH − CH3 ⎯KCN⎯ →⎯⎯ CH3 − CH − OCOCH3 | CN Несмотря на то, что выход акрилонитрила при термическом разложении ацетата лактонитрила достигал 85%, а степень превращения 50% [101,102], метод по эффективности все же уступал методу прямого получения из окиси этилена. В промышленности этот метод, как и способ прямой дегидратации лактонитрила (рис.5) широкомасштабного использования не получил. В Германии впервые появились установки получения акрилонитрила на основе окиси этилена. Уже было отмечено, что в 40-х годах в той же Германии создается метод получения акрилонитрила прямым гидроцианированием ацетилена. В 1930 г. Баум и Герман, пропуская смесь ацетилена с синильной кислотой над активированным углем, пропитанным Ba(CN)2 при температуре 400-500°С, получили 10% акрилонитрила. Затем Куру в качестве катализатора использовал смесь одного моля хлорида меди и 0,8 моля хлористого аммония. Построенные в тот период промышленные установки работали и в жидкофазном, и в газофазном режиме. Но оказалось, что в паровой фазе, осуществляя пропускание смеси равных объемов ацетилена и синильной кислоты при 400-500 oС над цианидами щелочных и щелочноземельных металлов, хлоридом меди, активированными тонкоизмельченными цинком, кадмием или их окислами, окисью алюминия, бокситом и т.д., наряду с акрилонитрилом образуется значительное количество побочных продуктов, что и объяснило применение в промышленности в основном жидкофазного гидроцианирования ацетилена (рис.6). В схеме на рисунке 6 ацетилен и синильную кислоту пропускали через жидкий слой катализатора, загруженного в реактор (CuCl, KCl, KCN, NaCl, HCl, H2O). 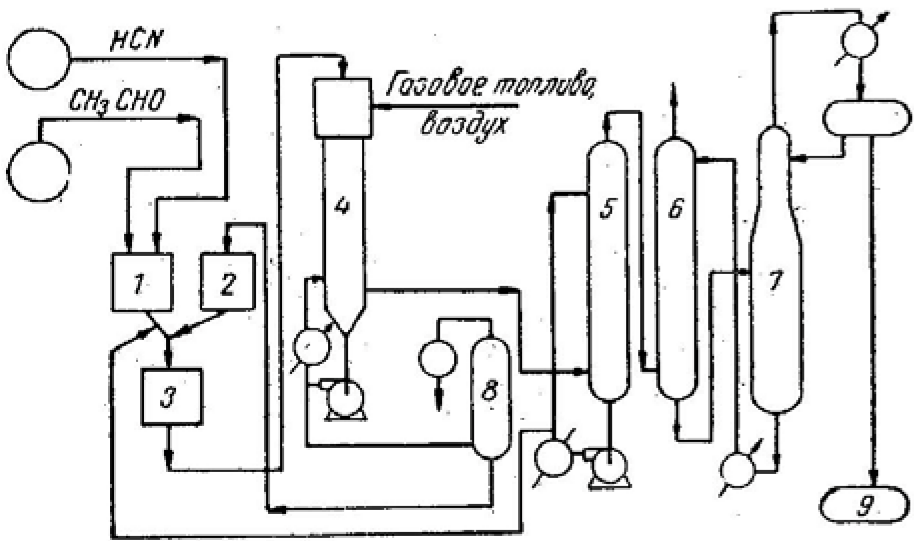 Рис. 5. Схема процесса производства акрилонитрила из ацетальдегида и синильной кислоты: 1 – аппарат для получения лактонитрила; 2 – емкость фосфорной кислоты; 3 – смеситель; 4 – реактор дегидратации лактонитрила; 5,6,7 – система очистки акрилонитрила; 8 – аппарат концентрирования фосфорной кислоты; 9 – емкость акрилонитрила. 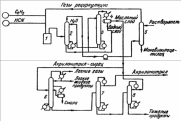 Рис.6. Схема процесса производства акрилонитрила из ацетилена и синильной кислоты: 1 – реактор; 2 – абсорбер; 3 – стриппер; 4 – сепаратор; 5 – скруббер; 6 – колонна разделения легких продуктов; 7 – колонна разделения продуктов; 8 – колонна разделения тяжелых продуктов. Для уменьшения образования побочных продуктов к катализаторам добавляли такие соединения как HgCl2, ZnCl2, BiCl3, As2O3, Bi2O3, SbO2. Реакция осуществлялась при 90°С и давлении 1,52,5 атм. Газообразные продукты реакции поступали в абсорбер, внизу которого находился отвод для водного слоя акрилонитрила, вверху – ацетилен и дивинилацетилен. Процесс был разработан американской компанией «Американ Цианамид Компани», но надо отметить, что очень большим недостатком процесса было образование таких продуктов как винил- и дивинилацетат, изопрен, 2-цианбутадиен, которые полимеризовались. Поэтому к недостаткам процесса добавлялась также трудность выделения и очистка целевого продукта. Однако, в Японии успешно использовался метод гидроцианирования ацетилена в псевдосжиженном слое катализатора с высокой избирательностью, при конверсии ацетилена 92%. В 50-е годы ХХ века был предложен и еще один метод синтеза акрилонитрила – метод окислительного аммонолиза пропилена. Первой работой по исследованию этого метода синтеза была работа Синклера. Пропилен и его производные были основным сырьем для синтеза акрилонитрила, потребность в котором с 1950 г. существенно возросла, так как увеличилось производство синтетических волокон на основе его полимеров и сополимеров. Фирмой Дюпон был разработан процесс получения акрилонитрила окислением пропилена окислами азота на серебряном катализаторе, промотированном окисью кальция в температурном интервале 420-500°С. Именно в тот период появление в больших количествах производных пропилена: акролеина, акриловой кислоты, аллилового спирта было очень актуальным. Окисление пропилена увеличило возможность вовлечения акролеина в производство акрилонитрила и глицерина по бесхлорному методу: CH2 = CH − CHO + NH3 +1/ 2O2 → → CH2 = CH − CN + 2H2O Температура процесса 250-600oС, время контакта 0.1-10 с. В качестве активной массы катализатора были рекомендованы элементы: Cu, Cr, W, Mn, Fe, Ca, Ni, Mo, Ag, Zn, Cd, Pb, Bi, P, Co и др. и их соединения, нанесенные на силикагель, силикаты, пемзу, каолин, окись алюминия, кизельгур и др. Наиболее перспективными оказались катализаторы на основе молибдена и его соединений. Достаточно прогрессивным способом получения акрилонитрила оказался прямой синтез на основе пропилена, аммиака и кислорода воздуха, осуществляемый в реакторах проточного типа со стационарным и псевдосжиженным слоем катализатора. Этот процесс был разработан фирмой Стандарт Ойл еще в 1959 году. В качестве катализатора процесса был выбран висмутмолибдено-фосфорный катализатор. Принципиальная схема получения акрилонитрила окислительным аммонолизом пропилена представлена на рис.7. 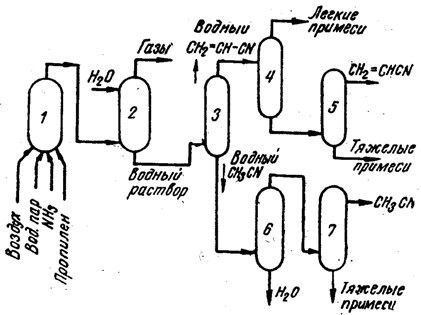 Рис. 7. Принципиальная схема получения акрилонитрила окислительным аммонолизом пропилена: 1 – реактор с кипящим слоем катализатора; 2 – колонна газоразделения; 3 – колонна разделения продуктов; 4,5,6,7 – ректификационные колонны системы очистки. ЗаключениеВ Советском Союзе процессы окислительного аммонолиза разрабатывались во ВНИИОлефин г. Баку акад. Далиным М.А., акад. Мехтиевым С.Д., проф. Серебряковым Б.Р., проф. Мехтиевым С.И., Расулбековой Т.И., Коновальчуковым А.В. и др. Исследования в области реакций нитрилов проводились в Горьковском политехническом институте и его филиале в г. Дзержинске проф. Зильберман Е.Н. На опытных заводах этих предприятий были внедрены процессы производства акрилонитрила и метакрилонитрила на основе пропилена и изобутилена. Тем не менее в работах представлены катализаторы одностадийного процесса окислительного аммонолиза пропилена в акрилонитрил в присутствии фосфорно-молибденовых или фосфорно-вольфрамовых солей висмута, сурьмы или олова, осажденных на силикагеле. В работе английских ученых для окислительного аммонолиза в качестве катализатора предложен арсеномолибдат висмута, нанесенного на силикагель, а фирмой Дистиллерс Компани Лимитед запатентован кобальто-молибденовый и сурьмянооловянный катализатор. Фирмой Стандарт Ойл был разработан процесс для двух японских компаний Асахи Кемикл Индастри и Мицубиси Кемикл Индастри. Обычно процесс осуществляется в псевдосжиженном слое катализатора при температуре 400-500°С, времени контакта несколько секунд и давлении 0.5-2 атм. Японская фирма Асахи предложила способ получения акрилонитрила из газовых смесей пропилена, аммиака и воздуха при температуре 300-480°С в присутствии катализатора, содержащего вольфрам, окись вольфрама, фосфорную кислоту и теллур или окись теллура, а также молибдат Co. Анализируя процесс аммонолиза можно утверждать, что проблема выбора катализатора из широкого ассортимента предлагаемых заслуживает особого аналитического подхода. Список использованной литературыРоговин З.А. Основы химии и технологии химических волокон. М.: Химия, 1974. Т.1,2. Юркевич В.В., Пакшвер А.Б. Технология производств химических волокон. М.: Химия, 1987. 304 с. Зазулина З.А., Дружинина Т.В., Конкин А.А. Основы технологии химических волокон. М.: Химия, 1985. 343 с. Карбоцепные синтетические волокна/Под ред. "К.Е.Перепелкина. М.: Химия, 1973. 589 с. Папков СП. Теоретические основы производства химических волокон. М.: Химия, 1990. 390 с. Зябицкий А. Теоретические основы формования волокон. М.: Химия, 1979.504 с. Симамура С. Углеродные волокна. М: Мир, 1987. 278 с. Конкин А.А. Углеродные и другие жаростойкие волокнистые материалы. М.: Химия, 1974. 168 с. Устинова Т.П. Зайцева Н.Л. // ПАН-волокна: технология, свойства, области применения. Саратов. 2003. |