Дерматополимиозит реферат. Введение Эпидемиология
 Скачать 0.72 Mb. Скачать 0.72 Mb.
|

|
Содержание
Введение Дерматополимиозит (ДМ) (болезнь Вагнера, болезнь Вагнера-Унферрихта-Хеппа) – тяжелое системное прогрессирующее заболевание соединительной ткани, характеризующееся аутоиммунным воспалительным поражением мышц, кожи и сосудов микроциркуляторного русла с повреждением внутренних органов, которое относят к группе идиопатических воспалительных миопатий. У 25-30% пациентов кожный синдром отсутствует. В этом случае говорят о полимиозите (ПМ). Ни ПМ, ни ДМ не имеют какой-либо четко определенной клинической картины. Поэтому долгое время считалось, что ПМ и ДМ – это одно и то же заболевание. В настоящее время установлено, что это различные болезни, несмотря на сходство отдельных симптомов. Эпидемиология Распространенность ДМ составляет 9-11 случаев на 100 тыс. населения. Среди пациентов преобладают женщины (около 70%). Патологию выявляют во всех возрастных группах. Отмечено два возрастных пика заболеваемости ДМ: 50 лет у взрослых и 10-15 лет – у детей. В старших возрастных группах ДМ может ассоциироваться с опухолевым процессом. Этиология Причины возникновения заболевания окончательно не выяснены. В настоящее время ДМ рассматривают как мультифакторный процесс с генетической предрасположенностью. Наибольшее значение придается инфекционным агентам, которые при определенных условиях, например, вследствие недостаточности супрессорных факторов, провоцируют аутоиммунное воспаление. В качестве этиологически значимых рассматривают вирусы гриппа, парагриппа, гепатита В, пикорнавирусы, парвовирус, а также простейшие (токсоплазма). Среди бактериальных возбудителей подчеркивается роль боррелиоза и β-гемолитического стрептококка группы А. В то же время прямое повреждающее влияние вирусов и других инфекционных агентов на мышцы не доказано, как и не доказана патогенетическая роль образующихся антимышечных АТ. На роль инфекционных факторов косвенно указывает более частое начало заболевания зимой и ранней весной, что по времени совпадает с увеличением числа инфекционных заболеваний. Несомненную роль играет генетическая предрасположенность, хотя заболевание не относится к наследственной патологии. О генетической предрасположенности свидетельствует нередкое развитие ПМ и ДМ у монозиготных близнецов и кровных родственников больных. Обнаруживается ассоциация заболевания с носительством антигенов HLA-B8 и DR3. Это тесно связано не с самим заболеванием, а с определенными иммунными нарушениями и в первую очередь с гиперпродукцией миозинспецифических аутоантител. Давно отмечена высокая частота сочетания ДМ (при остром или подостром его течении) со злокачественными опухолями – до 50% всех случаев. Это является особенностью ДМ, которая не повторяется или повторяется значительно реже при СКВ или системной склеродермии. Чаще выявляются опухоли легкого, желудка, молочных желез, кишечника, простаты, яичников. Считается, что ДМ у лиц старше 60 лет почти всегда имеет опухолевое происхождение (вторичный, паранеопластический ДМ). Провоцирующими факторами для первичного (идиопатического) ДМ могут быть введение вакцин, сывороток (противостолбнячной, противодифтерийной, БЦЖ), прием лекарств (антибиотиков, сульфаниламидов и т.д.), контакт с химическими агентами (эпоксидные смолы), травмы, хирургическое вмешательство, инсоляция, переохлаждение, эмоциональный стресс, гормональная перестройка (климакс, беременность, роды). Патогенез Основную роль в патогенезе ПМ и ДМ играют клеточные иммунные реакции. Основным патогенетическим фактором, вызывающим иммуновоспалительный процесс в мышцах, соединительной ткани, коже, вероятно, служат аутоантитела, хотя некоторые авторы считают, что они являются лишь свидетелями процесса. Механизм их образования до сих пор не расшифрован. При данной патологии выявляются аутоантитела против цитоплазматических белков и рибонуклеиновых кислот мышечной ткани, антитела к аминоацетилсинтетазам тРНК (аминоацетилсинтетаза катализирует связывание отдельных аминокислот с соответствующей тРНК), Jо-1-синтетазам, к Мi-2 (белково-ядерному комплексу с неизвестной функцией), к фактору I-а (обеспечивающему перенос аминоацил-тРНК к рибосомам), к SRP-антигену. Под влиянием вируса и генетической предрасположенности или опухолевых антигенов происходит нарушение (дисрегуляция) иммунного ответа, выражающееся в дисбалансе В- и Т-системы лимфоцитов: в организме вырабатываются антитела к скелетным мышцам и развивается сенсибилизация Т-лимфоцитов к ним. Реакция «антиген-антитело» и цитотоксический эффект сенсибилизированных к мышцам Т-лимфоцитов способствуют образованию и отложению в мышцах и микроциркуляторном русле различных органов иммунных комплексов. Их элиминация приводит к высвобождению лизосомных ферментов и развитию иммунного воспаления в мышцах и внутренних органах. При воспалении высвобождаются новые антигены, способствующие дальнейшему образованию иммунных комплексов, что ведет к хронизации заболевания и вовлечению в патологический процесс ранее здоровых мышц. 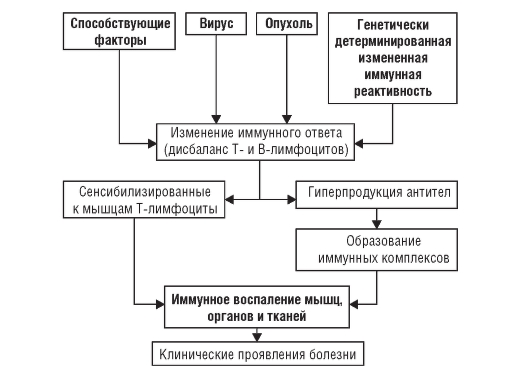 Между ПМ и ДМ выявлены определенные иммунопатологические различия. При ДМ в составе мышечного инфильтрата преобладают CD4- Т-лимфоциты, макрофаги и В-лимфоциты, а при ПМ – цитотоксические CD8- Т-лимфоциты. Предполагают, что при ДМ развивается гуморальный иммунный ответ, приводящий к активации комплемента и поражающий внутримышечные сосуды мелкого калибра, а при ПМ преобладают клеточные цитотоксические реакции, опосредуемые CD8- Т-лимфоцитами, синтезирующими, цитотоксические субстанции (перфорин). Патогенетическое значение миозитспецифических аутоантител не доказано. Важное значение в формировании и поддержании воспалительных реакций в соединительной ткани имеет выработка мононуклеарными клетками брадикинина, способствующего повышению проницаемости сосудов микроциркуляторного русла, продукции эндотелиальными клетками вазоактивных соединений (гистамина, простагландинов и др.). Происходит активация тромбоцитов и лимфоцитов, сопровождающаяся синтезом широкого спектра провоспалительных цитокинов, проникающих в окружающие сосуды ткани и способствующих развитию воспалительного процесса вокруг кровеносных сосудов с последующей перифасцикулярной атрофией. Считается, что поражение капилляров (микроангиопатия) поперечно-полосатых мышц комплексами АГ-АТ-комплемент лежит в основе развития ДМ. При этом имеется морфологическая картина, характерная для васкулитов. Для ПМ в большей степени присуща инфильтрация пораженных мышц цитотоксическими Т-лимфоцитами и повышение на них экспрессии молекул гистосовместимости, т.е. максимально активируется клеточный, а не гуморальный компонент иммунной системы. Варианты ДМ: - идиопатический (первичный); - паранеопластический (вторичный, опухолевый); - ювенильный (детский); - ДМ (ПМ) в сочетании с другими диффузными болезнями соединительной ткани. Классификация Общепринятой классификации ДМ не существует. Классификация Е. М. Тареева, Н. Г. Гусева (1965) Происхождение: I. Идиопатический (первичный). II. Паранеопластический (вторичный — составляет 20–30 % всех случаев). Течение: I. Острое. II. Подострое. III. Хроническое. Периоды: I. Продромальный (от нескольких дней до месяца). II. Манифестный с кожным, мышечным, общим синдромами. III. Дистрофический, или кахектический, терминальный, период осложнений. Степени активности: I, II и III. Основные клинические синдромы (признаки). По течению выделяют 3 основные формы: острую, подострую и хроническую. Острое течение характеризуется генерализованным поражением мускулатуры вплоть до полной обездвижимости, дисфагией, эритемой, поражением сердца и других органов с летальным исходом через 2–6 месяцев от начала ДМ. При своевременной массивной терапии глюкокортикоидами возможен переход к подострому и хроническому течению. Подострое течение характеризуется постепенным нарастанием симптомов, цикличностью, через 1–2 года от начала ДМ отмечается развернутая клиническая картина. Хроническое течение — более благоприятный вариант с умеренной мышечной слабостью, миалгиями, эритематозной сыпью, иногда без поражения кожи. О степени активности судят на основании неспецифических острофазовых показателей (увеличение СОЭ при 1 степени — до 20, при II степени — 21–40, при III степени — более 40 мм/ч) и иммунологических тестов, в частности, увеличения IgG. Клинические проявления Начало заболевания обычно постепенное, но может быть и острым (у детей и лиц молодого возраста) с выраженными общими симптомами болезни (лихорадка, похудание и др.) и миалгиями. В последующем постепенно (в течение нескольких недель) присоединяется прогрессирующая слабость в проксимальных группах мышц. Развернутая клиническая картина включает следующие проявления: - Кардинальным симптомом ДМ является симметричная слабость мышц плечевого и тазового поясов, сгибателей шеи и мышц брюшного пресса. Мышечная слабость нарастает постепенно, иногда сопровождается миалгиями. При остром начале ДМ выявляются распространенные отеки. Пациенты замечают затруднение в выполнении повседневных действий: подъем по лестнице, вставание с низкого стула, посадка в транспорт, умывание и причесывание. Походка становится неуклюжей, ковыляющей, больные не могут подняться без посторонней помощи с низкой поверхности, повернуться в кровати и оторвать голову от подушки. Прогрессирование заболевания приводит к неспособности удерживать голову в вертикальном положении вплоть до полного свисания головы. Поражение поперечно-полосатых мышц глотки, гортани и верхней трети пищевода приводит к дисфонии, дисфагии. Вовлечение межреберных мышц и диафрагмы может привести к дыхательной недостаточности. У половины больных отмечают миалгии или болезненность мышц при пальпации, их отек, однако мышечные атрофии развиваются только у пациентов, длительно страдающих ПМ или ДМ, особенно при отсутствии адекватной терапии. Воспалительные изменения в мышцах сопровождаются нарушением их кровоснабжения, что приводит к уменьшению мышечной массы, разрастанию в мышцах соединительной ткани и развитию сухожильно-мышечных контрактур. - Поражение кожи – патогномоничный признак ДМ. Симптом Готтрона – появление красных и розовых, иногда шелушащихся узелков и бляшек на коже в области разгибательных поверхностей суставов (чаще межфаланговых, пястно-фаланговых, локтевых и коленных). Иногда симптом Готтрона представлен только неярким покраснением, впоследствии полностью обратимым. Гелиотропная сыпь – лиловые или красные кожные высыпания на верхних веках и пространстве между верхним веком и бровью (симптом «лиловых очков»), часто в сочетании с отеком вокруг глаз. Сыпь может располагаться также на лице, на груди и шее (V-образная), на верхней части спины и верхних отделах рук (симптом «шали»), животе, ягодицах, бедрах, голенях. Часто на коже у пациентов появляются изменения по типу ветки дерева (древовидное ливедо) бордово-синюшного цвета в области плечевого пояса и проксимальных отделов конечностей. Характерные кожные проявления, наблюдаемые не только при ДМ, но и при ПМ: шелушение и растрескивание кожи пальцев и ладоней («рука механика» или «рука ремесленника»), гипертрофия кутикулы, околоногтевая эритема, телеангиэктазии. Ранними признаками заболевания могут быть синдром Рейно и изменения сосудов околоногтевого ложа, выявляемые при капилляроскопии. Отмечают расширение и дилатацию капиллярных петель (покраснение околоногтевых валиков). Также присутствует разрастание кожи вокруг ногтевого ложа. Зачастую кожные проявления при ДМ предшествуют поражению мышц в среднем на несколько месяцев или даже лет. Изолированное поражение кожи встречается чаще, чем поражение мышц и кожи одновременно. - Поражение суставов - может предшествовать развитию мышечной патологии. Чаще всего вовлекаются мелкие суставы кистей, лучезапястные суставы, реже – локтевые и коленные. Поражение двустороннее, симметричное, напоминает таковое при ревматоидном артрите, как правило, имеет преходящий характер и быстро купируется при назначении ГК. Поражение суставов проявляется болями и утренней скованностью, ограничением подвижности. - Кальциноз мягких тканей (преимущественно мышц и подкожной жировой клетчатки) – возникает на поздних стадиях, является особенностью ювенильного ДМ, наблюдается в 5 раз чаще, чем при ДМ у взрослых, особенно часто в дошкольном возрасте. Кальциноз может быть ограниченным или диффузным, симметричным или асиммитричным. Представляет собой кальцификаты – отложения депозитов солей кальция (гидроксиапатитов) в коже, подкожной клетчатке, мышцах или межмышечных пространствах в виде единичных узелков, крупных опухолевидных образований, поверхностных бляшек. При поверхностном расположении кальцинатов возможна воспалительная реакция окружающих тканей, нагнаивание и отторжение их в виде крошковатых масс. Глубоко расположенные кальцинаты мышц, особенно единичные, доступны для выявления только при рентгенологическом исследовании. - Поражение легких – частое поверхностное дыхание, инспираторная одышка, развитие гипостатической пневмонии. Одышка и кашель, хрипы и крепитация наблюдаются при выраженном поражении легких. Возможно развитие острого диффузного альвеолита – непродуктивный кашель и быстропрогрессирующая ДН. Чаще наблюдают медленное прогрессирование интерстициального легочного фиброза, который у некоторых больных определяют только при специальном исследовании. В наиболее тяжелых случаях развивается аспирационная пневмония. - Поражение сердца – в большинстве случаев протекает бессимптомно. Иногда при специальном исследовании находят нарушения ритма и проводимости (тахикардию, аритмию). В некоторых случаях развиваются очаговый или диффузный миокардит, дистрофические изменения миокарда, явления кардиосклероза, иногда с нарушением ритма. - Поражение ЖКТ – признаки гастроэнтероколита, причиной которых является распространенное поражение сосудов с развитием нарушений трофических процессов слизистой оболочки, ухудшением нервной проводимости и поражением гладкой мускулатуры. - Поражение почек – редко, хотя возможно развитие протеинурии и даже нефротического синдрома. Выраженная миоглобинурия может приводить к ОПН. Лабораторная и инструментальная диагностика Лабораторные исследования: - ОАК: патогномоничные для данного заболевания проявления отсутствуют. У части пациентов отмечается умеренная анемия, лейкоцитоз с нейтрофильным сдвигом влево, реже – лейкопения, эозинофилия. СОЭ увеличивается соответственно активности патологического процесса. Снижение титра комплимента, в небольшом количестве — LE-клетки, антитела к ДНК, снижение количества Т-лимфоцитов и Т-супрессорной функции, повышение содержания IgМ и IgG и снижение IgА. - ОАМ: повышение креатинина, миоглобина. - БАК: повышение активности КФК-МВ (превышение нормы в 5-50 раз, за исключением случаев тяжелой мышечной атрофии, дебюта ДМ, опухолевого миозита), повышение активности трансаминаз (АЛТ, АСТ, ЛДГ, альдолазы – неспецифично), повышение α2- и γ-глобулинов, миоглобина, гаптоглобина. - Иммунологические исследования: основное значение имеет определение миозит-специфических аутоантител: анти-Jo-1 (наиболее характерны для ПМ), анти-Мi-2 (наиболее характерны для ДМ). Однако названные антитела обнаруживаются только у 40-50% пациентов. - Исследование биоптатов кожно-мышечного лоскута: некроз мышечных волокон, потеря поперечной исчерченности, фрагментация и вакуолизация мышц, их атрофия и фиброз; лимфоцитарная и гистиоцитарная инфильтрация. В коже – атрофия сосочков, дистрофия волосяных фолликулов и сальных желез, изменения коллагеновых волокон, инфильтрация (при ПМ преимущественно в фасции, при ДМ – периваскулярная). Инструментальные исследования: - Игольчатая электромиография: характерны выраженные мышечные нарушения – короткие волны с полифазовыми изменениями, фибриллярные осцилляции в состоянии покоя. Используется как метод контроля эффективности лечения. - Электрокардиография: нарушения ритма и проводимости. - Рентгенологическое исследование: позволяет визуализировать структуру мягких тканей. В острой стадии ДМ мышцы выглядят более прозрачными, отмечаются просветления. При хроническом ДМ появляются кальцификаты в мягких тканях. В костях может быть умеренный остеопороз. На рентгенограмме грудной клетки определяется интерстициальный фиброз легких, преимущественно базальных отделов, кальцификаты плевры. Сердце увеличено в размерах. Более чувствительным методом считают КТ. - Спирография: рестриктивный тип дыхательной недостаточности – уменьшение величины жизненной емкости легких (ЖЕЛ) при нормальном объеме форсированного выдоха за 1с (ОФВ1). Индекс Тиффно – 70% или больше нормы. Диагностические критерии ДМ/ПМ: • основные: - характерное поражение кожи: периорбитальный отек и эритема (симптом «очков»); телеангиэктазии, эритема на открытых участках тела (лицо, шея, верхняя часть груди, конечности); - поражение мышц (преимущественно проксимальных отделов конечностей), выражающееся в мышечной слабости, миалгиях, отеке и позже – атрофии; - характерная патоморфология мышц при биопсии (дегенерация, некроз, базофилия, воспалительные инфильтраты, фиброз); - увеличение активности сывороточных ферментов – КФК, альдолазы, трансаминаз на 50% и более по сравнению с нормой; - характерные данные электромиографического исследования; • дополнительные: - кальциноз; - дисфагия; • диагноз ДМ достоверен: - при наличии 3-х основных критериев и сыпи; - при наличии 2-х основных, 2-х дополнительных критериев и сыпи; • диагноз ДМ вероятен: - при наличии первого основного критерия; - при наличии 2-х остальных из основных критериев; - при наличии одного основного и 2-х дополнительных критериев; • диагноз ПМ достоверен: - при наличии 4-х критериев без сыпи. Дифференциальная диагностика Дифференциальную-диагностику ИВМ проводят с широким кругом заболеваний, сопровождающихся проксимальной мышечной слабостью. - неврогенные миопатии (амиотрофический склероз, полиневропатия, спинальная амиотрофия Кугельберга-Веландера, синдром Кеннеди (спинобульбарная мышечная атрофия передних рогов спинного мозга), демиелинизирующие полинейропатии (острая, хроническая), невральная перонеальная амиотрофия Шарко-Мари-Тута); - первично-мышечные заболевания: инфекционные миозиты (бактериальные и вирусные миозиты), поражение мышц при токсоплазмозе, трихинеллезе, цистицеркозе, эхинококкозе; - лекарственные миопатии могут возникать при использовании ГК, пеницилламина, хлорохина (например, делагил), гидроксихлорохина (например, плаквенил), колхицина, статинов, гемфиброзила, эритромицина, эметина, зидовудина, а также при алкогольной и наркотической (кокаин) интоксикации, длительном приѐме гормонов щитовидной железы в высоких дозах. Для стероидной миопатии характерны нормальный уровень КФК, увеличение мышечной силы на фоне снижения дозы ГК; - метаболические миопатии (нарушение метаболизма гликогена, липидов, пуринов). Характерный признак-снижение толерантности к физической нагрузке и восстановление мышечной силы на фоне отдыха; - эндокринные миопатии; - прогрессирующие мышечные дистрофии; - рабдомиолиз и др. миопатии. Лечение - Следует рекомендовать пациентам избегать факторов, которые могут спровоцировать обострение болезни: отказаться от пребывания на солнце, от курения, от контактов и инфекционными больными, избегать физических и психо-эмоциональных перегрузок; - Следует рекомендовать пациентам исключить факторы, повышающие риск развития побочных эффектов терапии ГК: не употреблять в пищу сладкие продукты, включая мед и сладкие фрукты, повышающие риск развития стероидного сахарного диабета, также, исключение острой пищи, применение гастропротекторов с целью предотвращения язвенных осложнений (уровень доказательности С); - Все пациенты нуждаются в активной профилактике и лечении глюкокортикоидного остеопороза. Подбор антиостеопоретической терапии зависит от результатов денситометрического исследования и оценки дополнительных факторов риска остеопороза (менопауза, эндокринные заболевания). В зависимости от исходных данных минеральной плотности костной ткани назначаются препараты кальция в сочетании с витамином Д, или эти же препараты в сочетании с бисфосфанатами; - У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых, затрагивая мышечную ткань, может способствовать как формированию постинъекционных кальцинатов, так быть причиной ложноположительных результатов уровня креатинфосфокиназы (КФК). Программа терапии включает: - иммуносупрессивные средства (ГКС, цитостатики, циклоспорин А); - ВВИГ (внутривенный иммуноглобулин). Цель терапии – достижение регресса клинических проявлений. ГКС – основная патогенетическая терапия ДМ. Раннее начало терапии (в течение первых 3-х месяцев от начала симптомов) ассоциируется с благоприятным прогнозом. Используется пульс-терапия метилпреднизолоном или преднизолоном внутривенно капельно в течение трех суток или пероральный прием преднизолона в стандартных дозах. В зависимости от тяжести заболевания доза ГКС колеблется от 1 до 2 мг/ (кг•сут). Первые недели ее следует принимать в 3 приема в течение дня, а затем однократно в утренние часы. Улучшение состояния больных ПМ и ДМ развивается в среднем через 1-3 мес. Отсутствие положительной динамики в течение 4 нед считают основанием для увеличения дозы ГКС. После достижения клинического эффекта (нормализация мышечной силы и КФК) пациент продолжает получать индивидуально подобранную поддерживающую дозу стероидов. Дозировку снижают ежемесячно на 25% суточной дозы. Снижение дозы необходимо проводить под клиническим и лабораторным контролем. Вопрос об отмене ГКС решают при отсутствии клинико-биохимической активности процесса (активность КФК), исчезновении спонтанной активности по данным электромиографии в течение 12 месяцев. Дополнительно к ГКС назначают ВВИГ, что позволяет повысить эффективность проводимой терапии, в более ранние сроки начать снижение дозы ГКС. Применение ВВИГ 2 г/кг 1 раз в месяц в течение 3 месяцев является эффективным методом лечения ПМ/ДМ, резистентного к стандартной терапии. Потенциальным показанием для ВВИГ является тяжелая дисфагия (уровень доказательности В). Лечение ВВИГ способствует повышению общей сопротивляемости организма пациентов и более быстрому разрешению интеркуррентных инфекций. При резистентности к максимально высоким дозам ГКС или наличии сопутствующих аутоиммунных заболеваний используется монотерапия следующими иммуносупрессивными препаратами: метотрексат — 7,5–25 мг/неделю внутрь, подкожно или внутривенно; или циклоспорин А (сандиммун) — 2,5–3,5 мг/кг/сут; или азатиоприн — 2–3 мг/кг/сут. При недостаточной эффективности возможно сочетание нескольких иммуносупрессивных препаратов или их комбинация с ГКС. Общие принципы лечения иммуносупрессивными препаратами: - титрование дозы: назначение с небольшой дозы и постепенное ее повышение под контролем переносимости; - контроль переносимости: оценка уровня гемоглобина, числа лейкоцитов, тромбоцитов, азота мочевины, креатинина, активности АСТ, АЛТ. При уменьшении числа лейкоцитов менее 2,5х109/л и/или тромбоцитов – менее 100 х 109/л и повышении концентрации АСТ, АЛТ более чем в 3 раза от верхней границы нормы, лечение необходимо прекратить до устранения симптомов токсичности; - при присоединении интеркуррентной инфекции, в т.ч. герпетической – временная отмена иммуносупрессивных препаратов до исчезновения ее признаков. Плазмаферез используют у больных с тяжелым, резистентным к другим методам лечения полимиозитом/дерматомиозитом в сочетании с ГК и иммуносупрессивной терапией. Реабилитационные мероприятия и обучение пациентов Проводятся в зависимости от стадии заболевания: - В острую фазу противопоказаны ЛФК и физические нагрузки, проводимые пациентами «через силу»; допускаются только пассивные упражнения; - В стадию выздоровления - изометрические, а затем изотонические упражнения; - В хронической стадии - анаэробные упражнения. Профилактика При вторичных ДМ необходимо лечение основного заболевания (опухоль, трихинеллез). Первичная профилактика не проводится. Вторичная профилактика включает режимные ограничения (нагрузка, аллергизирующие факторы), диспансеризацию с проведением поддерживающей терапии глюкокортикоидами. Прогноз Выживаемость выше больных дерматомиозитом, чем при полимиозите. Для жизни при своевременной и адекватной терапии прогноз относительно благоприятный: пятилетняя выживаемость – 90%. К факторам, ухудшающим прогноз при ПМ и ДМ, относят: - пожилой возраст пациентов; - позднюю диагностику; - неадекватную терапию в начале болезни; - тяжелое течение миозита (лихорадку, дисфагию, поражение легких, сердца, ЖКТ); - миозит при злокачественных новообразованиях (5-летняя выживаемость – 50%). Клиническое наблюдение Больная Е., 64 года, госпитализирована в кожно-венерологическое отделение № 2/2 клиники кожных и венерических болезней (ККВБ) им. В.А. Рахманова 11.01.2016. При поступлении предъявляла жалобы на высыпания на коже волосистой части головы, лица, груди, шеи, разгибательной поверхности верхних конечностей и внутренней поверхности бедер, сопровождающиеся умеренной болезненностью и зудом. Семейный анамнез не отягощен. Сопутствующие заболевания: сахарный диабет 2-го типа, хронический холецистит, артериальная гипертензия II степени, экстирпация матки по поводу фибромиомы (1993г.). Анамнез заболевания: считает себя больной с октября 2012 г., когда впервые, на фоне активной инсоляции (во время 3-недельного пребывания в Австралии), отметила появление высыпаний на коже груди, лица и кистей. При обращении к дерматологу обсуждались диагнозы: системная красная волчанка, дерматомиозит, саркоидоз кожи. Была выполнена биопсия кожно-мышечного лоскута плеча с последующим гистологическим исследованием. Заключение: имеются некоторые черты, свойственные поражениям из группы коллагенозов (красной волчанки, дерматомиозита). Пациентка была направлена к ревматологу. Антинуклеарный фактор (АНФ) от 10.07.2013: 1/1280 (норма ‒ 1/160). Выставлен диагноз «кожная красная волчанка; АНФ+», назначено лечение плаквенилом (400 мг/сут в течение 2-х месяцев, далее поддерживающая доза 200 мг/сут в течение 2-х лет) ‒ без эффекта, процесс прогрессировал. В июле 2015 г. повторно обратилась к ревматологу. Выполнены анализы: ревмопробы от 18.07.2015: Антистрептолизин–О (АСЛ–О) ‒ отрицательный, Ревматоидный фактор (РФ) ‒ отрицательный, С-реактивный белок ‒ отрицательный. Назначен метилпреднизолон в дозе 4 мг/сут, который пациентка самостоятельно отменила через месяц в связи с отсутствием эффекта. 11.01.2016 г. обратилась в ККВБ им. В.А. Рахманова. При осмотре обратили на себя внимание: на коже плеч, груди, в зоне декольте, кистей, бедер – сливная, макулярная фиолетовая эритема, на поверхности которой отмечаются множественные телеангиэктазии; слегка отечная эритема кожи лица, особенно периорбитальной области с шелушением на поверхности; на коже волосистой части головы – шелушение и диффузное разряжение волос (рис. 1); в области проксимальных околоногтевых валиков пальцев рук ‒ телеангиэктазии; над межфаланговыми и пястнофаланговыми суставами, распространяющаяся линейно над сухожилиями разгибателей кисти и пальцев ‒ сливная макулярная розово-фиолетовая отечная эритема(рис.2). 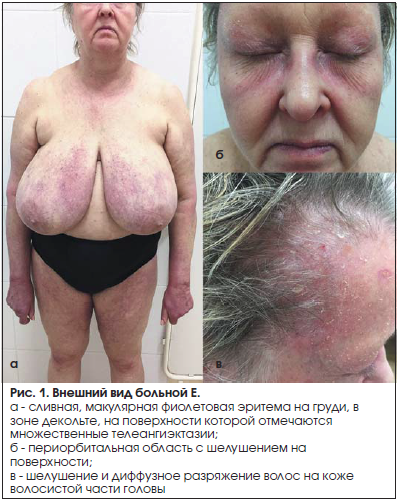 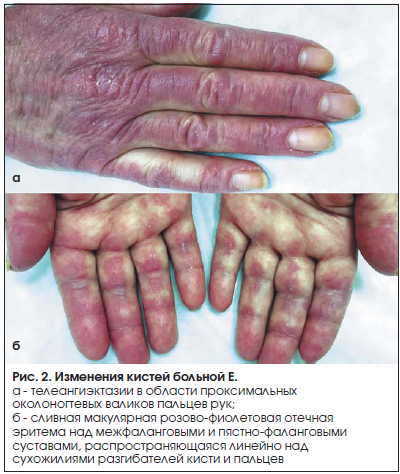 Проводился дифференциальный диагноз между амиопатическим дерматомиозитом и эритематозом. Проведено дополнительное лабораторно-инструментальное обследование. • Глубокая диагностическая биопсия подкожно-жировой клетчатки и прилегающей мышечной ткани в очаге в области плеча: ‒ результат гистологического исследования биоптата: эпидермис с очаговым уменьшением слоев, небольшой гиперкератоз, акантоз, дермоэпидермальный стык уплотнен, в дерме незначительные лимфомакрофагальные инфильтраты расположены периваскулярно или рядом с волосяными фолликулами. Заключение: изменения неспецифичны; ‒ данные иммуногистохимического исследования биоптата: Ig – умеренное накопление в сосочковом слое дермы (диффузно и гранулярно), в базальной мембране нет, распространенная фиксация в ядрах кератиноцитов всех слоев эпидермиса; IgM – незначительно в дермоэпидермальной зоне; IgA – следы в сосочковом слое дермы, в составе крупных гиалиновых телец; C3c-компонент комплемента ‒ незначительно в сосочковом и сетчатом слоях дермы; фибрин ‒ фиксация в сосудах дермы. Заключение: иммуноморфологическая картина не противоречит диагнозу «красная волчанка». •МСКТ органов грудной клетки: КТ-признаки нерезко выраженной лимфаденопатии внутригрудных лимфатических узлов. •ЭКГ: ритм синусовый. Умеренные изменения в миокарде. •ЭМГ: в обследованных мышцах признаки неактивного первичного мышечного процесса. •Денситометрия: показатели в пределах возрастной нормы. •Капилляроскопия: миопатический тип (наиболее часто подобные изменения встречаются при ДМ). •anti-CMV IgG: 616,1 Ед/мл (>= 6,0 ‒ положительно), anti-CMV IgM: отрицательно, anti-HSV (1-го и 2-го типа) IgG: 17,7 индекс позитивности (>1,1 ‒ положительно), anti-HSV (1-го и 2-го типа) IgM: отрицательно, anti-EBV IgG-EBNA (ядерный белок): 429 Ед/мл (>20 ‒ положительно), anti-EBV IgM-VCA (капсидный белок): <10 Ед/мл. •Биохимический анализ крови: альбумин – 59,8%; α1 – 3,9%; α2 – 9,0%; β1 – 10,4%; γ – 16,9%; КФК общая – 94 ед/л; АСТ – 19 ед/л; АЛТ – 21 ед/л; ЛДГ – 375 ед/л; билирубин общий – 8,1 мкмоль/л; креатинин – 0,69 мг/дл; альбумин – 44,5 г/л; белок общий – 69,5 г/л; КА – 2,83; глюкоза – 8,6 ммоль/л; холестерин – 7,3 ммоль/л; триглицериды – 2,80 ммоль/л; ЛПНП – 4,14 ммоль/л; ЛПОНП – 1,27 ммоль/л; ЛПВП –1,91ммоль/л. Учитывая типичную клиническую картину, капилляроскопию, ЭМГ, был выставлен окончательный диагноз «амиопатический дерматомиозит» и проведено лечение: метилпреднизолон 24 мг/сут и курс лечебного плазмафереза № 5. На фоне лечения отмечался выраженный положительный эффект в виде регресса высыпаний на 70%. Литература
|