теория к колобу 2 патфиз (болезни крови). теория к КОЛОБУ 2. 13. нарушения тканевого роста. Опухоли. Механизмы канцерогенеза
 Скачать 2.86 Mb. Скачать 2.86 Mb.
|

|
13. НАРУШЕНИЯ ТКАНЕВОГО РОСТА. ОПУХОЛИ. Механизмы канцерогенеза Протоонкогены кодируют белки, регулирующие норм клеточный рост и дифф-ку Онкогены – мутанты протоонкогенов, стимулируют развитие рака HER-2/neu – кодирует рецептор клеточного деления Ras – транскрипция генов клеточного деления и пролиферации, мутированный Ras ↑пролиферацию (норм путь: клеточный рецептор – Ras – Raf – Erk – транскрипция; канцер: кл рецептор – Ras мутированный – Raf – Erk – усиленная пролиферация клетки). MYC – транскрипционный фактор, контроль экспрессии генов пролиферации. Src – белок тирозинкиназы, регулирует клеточную активность. Гены-супрессоры опухолевого роста p53 – транскрипционный фактор, активируется только при повреждении ДНК, останавливает деление клетки пока не восстановится ДНК. RB1 – ретинобластома, на 13 хромосоме, контроль клет деления, связ с транскрипц фактором. Гены репарации (восстановления) ДНК: аккуратное копирование каждой цепи ДНК во время деления клетки. • Мутации в генах репарации ДНК приводят к увеличению частоты мутаций в других генах (протоонкогенов и генов-супрессоров опух роста). • Гены участвующие в развитии рака груди (BRCA1 и BRCA2) • Гены участвующие в развитии наследуемого неполипозного рака кишечника (MSH2, MLH1, PMS1, PMS2) вовлечены в репарацию ДНК. • Мутации в этих генах вызывают опухолеобразование Антибластомная резистентность Антиканцерогенная:  Химических канцерогенов: инактивация эндогенных химических в-в (желчь, кал, моча), пино- и фагоцитоз канцерогенов, АТ против гаптенов/канцерогенов, ингибитор свободных радикалов антиоксидантами (вит А, бета-каротин, С, Е) – остановка СПОЛ, окисление оксидазами, восстановление редуктазами микросом, ферментативное и неферментативное деметилирование, ферментативная конъюгация с глюкуроновой/серной кислотой. Химических канцерогенов: инактивация эндогенных химических в-в (желчь, кал, моча), пино- и фагоцитоз канцерогенов, АТ против гаптенов/канцерогенов, ингибитор свободных радикалов антиоксидантами (вит А, бета-каротин, С, Е) – остановка СПОЛ, окисление оксидазами, восстановление редуктазами микросом, ферментативное и неферментативное деметилирование, ферментативная конъюгация с глюкуроновой/серной кислотой. Биологических (против онко-вирусов): ИФН (альфа синт-ся мф leu, бета – фибробластами и Т-л, гамма – NK и мф), АТ (блок проникн-е вируса в клетку). Физических (ион излуч): антирадикальные реакции (инактивация радикалов) и антиперекисные р-ии (инакт перекисей) – вит Е, селен, глутатион-дисульфидная система, глутатионпероксидаза, супероксиддисмутаза, каталаза. Антитрансформационная: Антимутационные механизмы: клеточные ферменты репарации ДНК (распознавание, разрезание рестриктазами, вставка норм уч ДНК, синтезируемого полимеразой). Антионкогенные механизмы: антионкогены (гены супрессоры опухолевого роста), подавляет размножение опух клетки, нормализует и стимулирует их дифф-ку. Антицеллюлярная (включается в момент образования первых бластом клеток). иммуногенные механизмы – NK-клетки, Т-киллеры, макрофаги и выделяемые ими ИФН и ФНО, АТ, неимунногенные – кейлоны (подавляют клеточное деление), гепарин. Предраковые состояния Предрак. Облигатный (полипоз желудка, толстой кишки, пигментная ксеродерма, нейрофиброматоз), факультативный (язвенная болезнь желудка, эрозия шейки матки, аноцидный гастрит). Стадии развития злокачественной опухоли Стадия неравномерной диффузной гиперплазии (предопухолевая стадия) – ткань норм строение, но увелич число кл/волокон; ограниченных участков пролиферации нет. Очаговая пролиферация – ткань норм строения, но в отдельных очагах бурная пролиферация. Относительно доброкачественная опухоль – возрастает клеточный и тканевой атипизм, пролиферации, БЕЗ инвазии и деструкции. Злокачественная опухоль – крайний атипизм, пролиферация, ИНВАЗИЯ, ДЕСТРУКЦИЯ (иногда злокач опухоль разв-ся сразу после 2 стадии). Профилактика и лечение опухолей Клиника: раннее выявление и лечение предопухолей, гормональных нарушений. Гигиена: борьба с загрязнением окр среды, с вредными привычками (алко, курение). Лечение: удаление опухоли (хирургически), уничтож опух клеток (химиотерапия, лучевая терапия). 14. ПАТОЛОГИЯ КРАСНОЙ КРОВИ ЭРИТРОЦИТОЗ Качественные изменения эритроцитов (нарушении созревания в КМ или ↑ проницаемости костно-мозгового барьера; изм типа кроветворения в КМ (с эритробластического на мегалобластический); насл и приобр нарушений обмена веществ, состава и структуры эритроцитов). Дегенеративные изм эритроцитов: Размер, анизоцитоз (микро- макроцит, мегалоцит, акантоцит – с острыми выпячиваниями при алко пораж печени, гипосплении; эхиноцит зубчатый при язвах раке желудка, уремии), Формы, пойкилоцитоз; окраски эритроцитов (гипо при железодефицит анемиях, гиперхромия – мегалобласт и макроцитарная анемия); наличии патологических включений в эритроцитах — телец Жолли (остаток ядра в виде1-3 глыбок), колец Кебота (остаток ядерн оболочка в виде кольца или 8), базофильной зернистости (агрегаты рибосом и митохондрий, при свинцовых и др интоксикация, мегалобласт сидеробластных анемиях, талассемии), тельца Гейнца - глыбки денатурированного Hb. Пойкило и анизоцитоз – В12 фолиево дефицитная анемия, железо дефицитная, насл гемолитическая анемия. Микросфероциты (без просветления в центре, мелкие) – насл гемолит анемия Минковского-Шоффара. Ретикулоциты (оценка регенераторной активности ККМ) Первичный эритроцитоз: б. Вакеза/эритремия (истинная полицитемия) – гиперплазия КМ, эритроидного ростка. Связана с опух пр-ссом (лейкоз) + поражение клетки предшественницы миелопоэза → неогранич пролиф-я этой клетки, дифф-ка по 3 росткам (миелоидному, эритроидному - преимущ, мегакариоцитарному). Лаб: ↑ эр-ты (7-10-12*1012/л), ↑ Hb, гипохромия, лейкоцитоз нейтроф сдвиг ядра влево, тромбоцитемия до 600-800*109/л, сниж СОЭ (1-2), вязкость повышена. Лечение: удаление массы эрит (эритроцитофорез), цитостатики, профилактика тромботич осложн-й: кровопускание, но с терапией с дезагрегантами. Вторичные эритроцитозы (следствие др заб-й). * Абсолютные (↑ эритропоэз в КМ) – ↑ эритропоэтина (гипоксия, ишемия, опухоли почек - эритропоэтинсинтезирующая). * Относительные – обезвоживание (сгущение крови), выброс крови из депо. (+классификация анемий по размеру, цв пок-лю, регенер спос, типу кроветв, течению По этиологии: насл и приобр, по п/г: см ниже таблица) АНЕМИИ
Патогенез гипопластической анемии 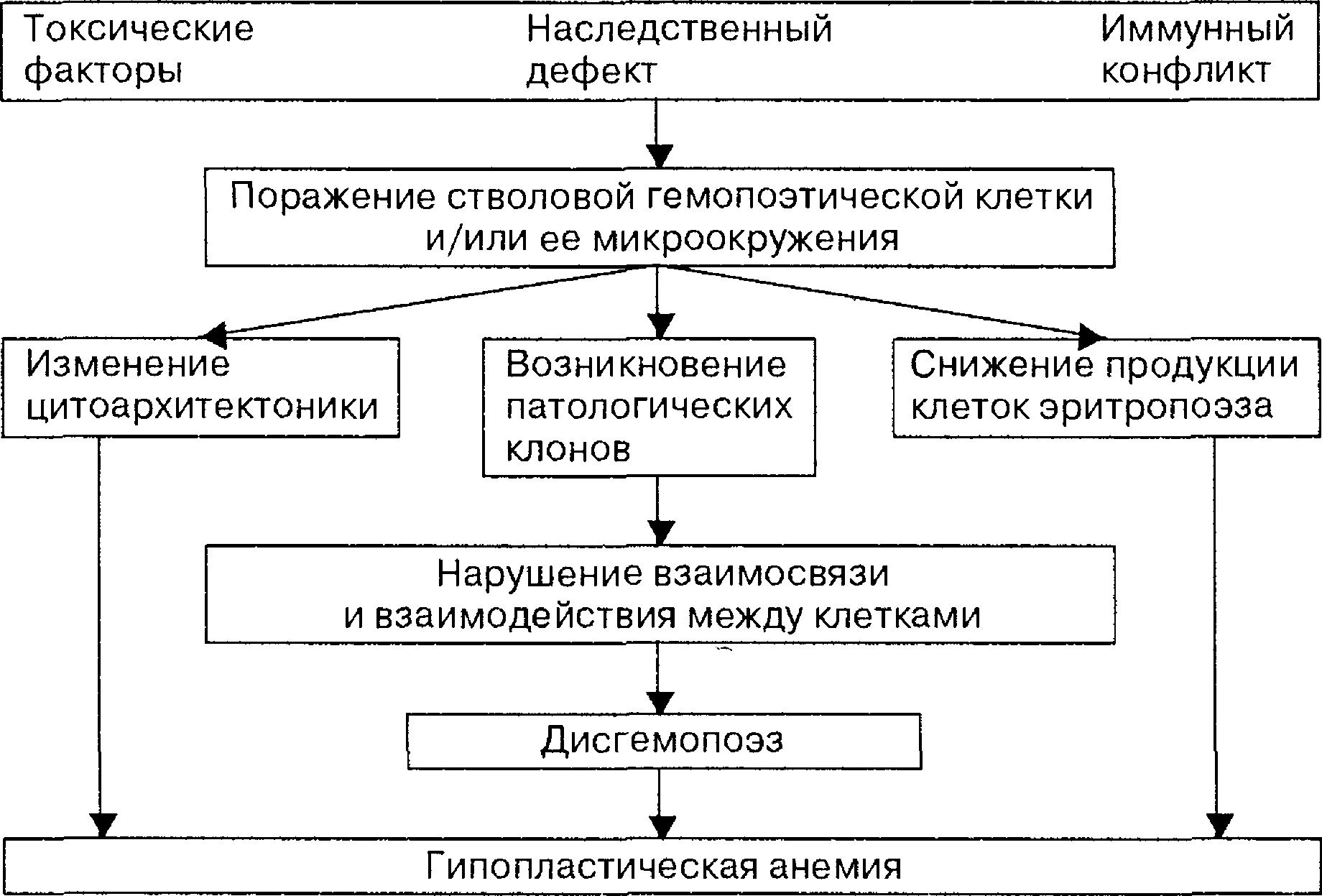    15. ЛЕЙКОЦИТОЗЫ. ЛЕЙКОПЕНИИ Патологические формы лейкоцитов 1. АНИЗОЦИТОЗ (микроформы или макрополициты – гигантские кл) 2. ПАТОЛОГИЯ ЯДРА Ней с гиперсегментацией(N 2-5 ней, 2-3 эоз) Причина: Нарушение биосинтеза нуклеиновых кислот (луч болезнь, ЛВ наруш с-з ДНК (гидроксилмочевина), в гигантских нейтрофилах при дефиците витамина В12 и фолиевой кислоты ("стареющие" клетки). Ней с гипосегментацией ядра. Насл доброкач аут-доминант, либо уменьш кол-ва сегментов, либо не до конца сегментирован (эллипс, гиря), либо полное отсут сегмента). При наследственных формах заболевания функция лейкоцитов сохраняется в норме. Приобретенные нарушения могут иметь место при лейкозах инфекциях, при действии лекарственных препаратов. Пикноз (уплотнен хроматит), хроматинолиз (разжижение хроматина), рексис (распад ядра на части), исчезн межсегмент нитей гранулоцитов, фрагментация яд хроматина (микроядра), лизис (растворение яд оболочки), вакуолизация (дырки в хроматине), голые ядра лимфоцитов (лимф «без цитоплазмы». Формы ядра: лимф с двудольчатым ядром, тени Боткина-Гумпрехта (раздавленные хрупкие лимфоциты – ХРОНИЧ ЛИМФОлейкоз). 3. ПАТОЛОГИЯ ЦИТОПЛАЗМЫ. Токсич зернистость. Грубая, темного цвета зернистость, появляется в цитоплазме в результате коагуляции белков при тяжелых инфекциях, интоксикациях. При этом в ней могут быть вакуоли. Вакуолизация цитоплазмы (пузырьки в цтплазме). Наблюдаются при сепсисе, тяжелых интоксикациях, инфекциях (может сочетаться с токсической зернистостью), семейной вакуолизации лейкоцитов (аномалии Джордана). Это признак жировой дегенерации клеток. Гранулоциты с кольцевыми ядрами. Образуются при хроническом алкоголизме. Лейкоциты с тельцами Князькова – Деле. В цтплзм круг/овал включения голубые. Обр-ся из РНК из фрагментов шероховатого ЭПС – признак дегенерации. Обнаруживаются при инфекционных заболеваниях в сочетании с токсической зернистостью или цитоплазматическими вакуолями. Палочки Ауэра – вишневого цвета (слипшиеся азурофильная зернистость). Лейкопении и лейкоцитозы – вторичны (показатель патологии) ЛЕЙКОПЕНИИ (абс и относит; физиол и патологич) Физиологическая (перераспределительная, и у 2% людей европеоидов как постоянный пок-ль). Патологическая: первичная (при сд Чедиака-Хигаси ленивые лейкоциты, семейная нейтропения, хронич гранулематоз); вторичная (ион излуч, ЛВ цитостатитики сульфаниламиды барбитураты, аутоиммунные, генерализ инфекции брюш тиф\паратиф). П/Г: нарушение лейкопоэза (генетич дефект, дефицит белков/В12/ФК, длит прием ЛВ); Чрезмерное разруш лейкоцитов (ион излуч, р/а, аутАТ к лейкоцитам, токсич в-ва); Перераспределение лейкоцитов (временно при шоке, тяжёлой, длительной мышечной работе, развитии феномена “краевого стояния” лейкоцитов (рожа, флегмона), при выходе большого количества лейкоцитов в ткани при их массовом повреждении (перитонит, плеврит, механическое повреждение мягких тканей); Повышенная потеря лейкоцитов (обширные ожоги, хронические гнойные процессы – остеомиелит, перитонит) ↓ снижается противоопухолевая и противоинфекционная резистентность ↓ генерализация септического процесса, инфицирование организма, могут развиться новообразования АГРАНУЛОЦИТОЗ (полное или почти полное отсут гранулоцитов <0,75*10 9/л или лейкоциты <1*10 9/л). Причина: Дей-е на КМ (цитостатики, антитиреоид средства, сульфаниламиды – стрептоцид, идиопатический). 1) миелотоксический – прямое воздей на КМ, сниж ф-ии КМ, из-за накопления ЛВ. 2) иммунный – гаптеновый, аутАТ, агглютинины, лизины против кл-предшеств (ЛВ) – клиника: некротич-язвенная ангинаю\. ЛЕЙКОЦИТОЗЫ Физиологический у здоровых новорожденных, при беременности, при физической нагрузке («миогенный»), пищеварении («пищеварительный»), при психических переживаниях («эмоциональный»), при смене часовых полюсов («акклиматизационный»). В большинстве физиол лейкоз – носит перераспределительный характер. Патологические лейкоцитозы вторичны, если убрать заболевание норма. 1. Усиление нормального лейкопоэза под влиянием лейкопоэтинов (инф, аллерг, аутоимм, ожог, отморожение, травма, инфаркт миокарда), кровотечениях, отравлениях, при облучении. 2. Перераспределение лейкоцитов в сосудистом русле (ложные, относительные лейкоцитозы). Может наблюдаться при травматическом, анафилактическом шоке, значительной физической нагрузке, при скоплении большого числа зрелых лейкоцитов в каком-либо органе и отсутствии признаков гиперплазии лейкопоэтической ткани, сохранении нормального числа лейкоцитов в крови. 3. Гиперпродукция лейкоцитов при опухолевом поражении гемопоэтической ткани (лейкозах) за счет пролиферации опух кл и стимуляции обр-я норм лейоцит вследствие появления в организме опухолевых АГ. 4. Гемоконцентрация. Её обусловливает гипогидратация организма с развитием гиповолемии (повторная рвота, диарея, полиурия). ЛЕЙКЕМОИДНЫЕ РЕАКЦИИ – усиленный лейкоцитоз (30-50*109/л), но НЕТ БЛАСТОВ, сильный сдвиг влево. НИКОГДА НЕ ПЕРЕХОДЯТ В ЛЕЙКОЗ. Этиология: колоние-стимулирующий фактор Типы: 1) миелоидный: а) промиелоцитарный (4 кл) – тяж токсикоинф, сальмонеллез, лекарст алл и алл дерматит, системная алл, ревматизм, повыш дозы преднезалона, спленэктомия, гемолитич анемия. Б) нейтрофильный – сепсис, острая кровопотеря, в сочет с токсикоинфекц. 2) лимфоцитарный – инф лимфоцитоз, ветрянка взрослых, иммунобластные лимфадениты (мононуклеоз, адено и энтеровирус инф, лекарств дерматиты, опояс лишай, коллагеноз, р-я трансплантат против хозяина). 3) моноцитарно-макрофагальный – туберкулез, болезни имм комплексов, идиопатич. 4) большие эозинофилии крови – паразит, опухоли, аллергоз, коллагеноз. 5) псевдобластные ЛР – только у новорожд при сд Дауна. У ЛР ВСЕГДА ЕСТЬ ПРИЧИНА (сепсис, гной инф, метастаз опухоли в КМ, имм и алл поврежд орг и тканей). Отличие от хронич лейкозов: есть причина (осн заб-е), меньше выражен сдвиг ядра по сравн с ХМЛ, не повышаются эоз и баз, дегенерация нейтрофилов, практически нет изм эритроцитов и тромбоцитов, лейк р-я проходит после лечения (лейкоз требует специф противоопух лечения). 16. ГЕМОБЛАСТОЗЫ – ТПП, опухоли из кроветворных клеток. ЛЕЙКОЗЫ – опухоли гемопоэтич ткани КМ (из лейкоцитарного или мегакариоцитарного ростка), есть бласты. ГЕМАТОСАРКОМА (из мезенхимальной СТ – попала в КМ – лейкемизация гематосаркомы) – опухоль гемопоэтич тк ВНЕ КМ. Признаки: местный локальный рост, их клетки первоначально не распред-ся по системе кроветворения. Эти 2 формы могут переходить одна в другую (метастазирование лейк кл за пределы КМ – гематосаркома, и наоборот). ЛИМФОЦИТОМА – опухоль кроветвор тк вне КМ, сост из зрелых лимфоцитов. ПРИЧИНЫ: физ (р/а), хим (длит интоксикация бензол-сод, цитостатики), биол (вирусы, ВИЧ – саркома Капоши, в. Эпшт-Барр – лимфома Беркитта, в.Т-клет лимфомы – лейкемии человека, вторично ВИЧ-СПИД) причина генетика, ИД, кахексии, голод. Лимфома Беркитта поражает поднижнечелюст ЛУ, растет опухоль быстро, через 6-12 нед ребенок =. Наследственность: сд Дауна (лейкозы в 18-20 р чаще), сд Шерш-Терн, сд Клайнф, Фалькони. Малые хромосом абберации (делеция транслокация филадельфийская хрмсма –ХМЛ), хронич семей лейкозы. Иммунные факторы: идс (луи бар, вискот-олдрич, брутон – острый лимфобластный лейкоз и лимфосаркома) и приобретен идс (цитостатики) в 35 р выше. 18. КОАГУЛОПАТИИ Тромбастения Гланцмана Развитие тромбоцитарной дисфункции обусловливается отсутствием или дефектом мембранного рецептора к фибриногену и гликопротеинам IIb – IIIa. Это приводит к резкому снижению интенсивности процесса связывания фибриногена с мембраной тромбоцита, в результате чего нарушается агрегация тромбоцитов.Заболевание наследуется аутосомно-рецессивно, проявляется уже в раннем детском возрасте, характеризуется петехиально-экхимозным типом кровоточивости, склонностью к кровотечению из слизистых оболочек (носовые, маточные кровотечения, кровоизлияния в склеру и сетчатку глаза), длительными кровотечениями после удаления зуба или ЛОР-операций. При исследовании семейного анамнеза в родословной выявляется пробанд больных родственников в семьях обоих родителей «по горизонтали». При лабораторной диагностике обнаруживается: увеличение времени кровотечения; нормальное количество тромбоцитов; в пределах нормы адгезия тромбоцитов, при изучении ристоцетининдуцированной агрегации тромбоцитов выявляется отсутствие типичной двухфазной кривой; АПТВ в норме. Синдром (болезнь) Бернара – Сулье (макроцитарная тромбоцитодистрофия, синдром гигантских тромбоцитов) При данной болезни в мембране тромбоцита отсутствует специфический гликопротеин, взаимодействующий с ФВ-VIII, ФV, ФIX и ристоцетином, а также повышается содержание сиаловых кислот, снижается электрический заряд. Это приводит к нарушению адгезионных свойств тромбоцитов. Болезнь наследуется аутосомно-рецессивно, характеризуется укорочением продолжительности жизни тромбоцитов при их нормальном процессе продуцирования в костном мозге, следствием чего является развитие умеренной тромбоцитопении. Основным морфологическим критерием заболевания является наличие в крови гигантских тромбоцитов, достигающих 6 – 8 мкм (в норме 2 – 4 мкм). Клиническая картина характеризуется кровоточивостью петехиального типа, тяжесть которой варьирует в больших пределах – от относительно легких и латентных форм до тяжелых и даже фатальных случаев. Тяжесть кровоточивости зависит от содержания аномальных тромбоцитов: чем выше их процент, тем тяжелее и потенциально опаснее протекает геморрагический синдром. При лабораторной диагностике определяются: увеличение времени кровотечения; тромбоцитопения, увеличение размера тромбоцитов; снижение адгезии тромбоцитов и ристоцетин-индуцированной агрегации; - нормальные показатели коагуляционного гемостаза в том числе АПТВ. Болезнь Виллебранда В основе развития заболевания лежит дефицит или функциональная неполноценность фактора Виллебранда (ФВ), наследуется заболевание аутосомнодоминантно с неполной пенетрантностью или (реже) – аутосомно-рецессивно. Дефицит и/или дефект ФВ приводит к нарушению процесса адгезии тромбоцитов к коллагену сосудистой стенки и снижению интенсивности образования комплекса ФВ – ФVIII, а также к уменьшению периода его полусуществования за счет ускорения катаболизма и элиминации ФVIII из крови. Клиническая картина болезни разнообразна, зависит как от фенотипического проявления патологического гена, так и от физиологического статуса организма (беременность, стресс, прием контрацептивов и т.д.). Дефицит и/или дефект ФВ приводит к нарушению как сосудисто-тромбоцитарного, так и коагуляционного гемостаза. Это проявляется экхимозными, реже – гематомными кровоизлияниями, меноррагиями, кровоточивостью слизистых оболочек. Характерен высокий риск профузных кровотечений при хирургических вмешательствах. При лабораторной диагностике устанавливается: увеличение времени кровотечения; нормальное количество тромбоцитов; снижение степени адгезии тромбоцитов к стеклу и ристоцетининдуцированной агрегации; снижение содержания и/или активности ФВ; - увеличение АПТВ. Наследственные коагулопатии – это заболевания, обусловленные дефицитом факторов VIII и IX, являются наиболее распространенными наследственными коагулопатиями (более 95% случаев). Дефицит факторов VII, X, V, XI составляет до 1,5%; дефицит других факторов (XII, II, I, XIII) встречаются крайне редко (в единичных случаях). Гемофилия А (дефицит фактора VIII) Заболевание наследуется рецессивно, сцеплённо с Х-хромосомой. Болеют лица мужского пола (10 случаев на 100 тыс. мужчин). Дефицит ФVIII приводит к резкому увеличению времени образования протромбиназного комплекса, что сопровождается длительным, практически не прекращающимся кровотечением при незначительной травматизации сосудов (прикусывание языка, ушибы и т.д.). Для гемофилии А характерен гематомный тип кровоточивости. Клиническая картина заболевания зависит от степени его тяжести, которая определяется мерой сохранения активности фактора VIII. При легкой форме заболевания (более 5% активности) кровотечения возможны лишь при значительных травмах или оперативных вмешательствах. Болезнь протекает субклинически и часто не диагностируется. При тяжелой или очень тяжелой форме (2% и менее 1%, соответственно) развиваются рецидивирующие кровоизлияния в крупные суставы (гемартрозы), приводящие к анкилозированию; крупные меж- и внутримышечные, забрюшинные гематомы с последующей деструкцией мягких тканей, тяжелые и частые спонтанные кровотечения, упорные рецидивирующие желудочно-кишечные и почечные кровотечения. При лабораторной диагностике выявляются: значительное увеличение АПТВ; ПВ и ТВ – в норме; нормальные показатели сосудисто-тромбоцитарного гемостаза (ВК, количество тромбоцитов в крови и др.); частичное или полное отсутствие активности ФVIII в плазме крови. Гемофилия В (болезнь Кристмаса, дефицит ФIX) Заболевание наследуется рецессивно, сцеплённо с Х-хромосомой. Данный дефект приводит к значительному замедлению формирования протробиназного комплекса, что обусловливает развитие кровоточивости гематомного типа. Клиническая картина гемофилии В также, как и гемофилии А, характеризуется кровотечениями (гемартрозы, гематомы), но частота их в 5 раз ниже, чем при дефиците ФVIII. Лабораторнаядиагностика свидетельствует, что: АПТВ увеличено, ПВ и ТВ в норме; показатели сосудисто-тромбоцитарного гемостаза в норме; - активность ФIХ частично снижена или отсутствует. Гемофилия С (дефицит ХI фактора) наследуется аутосомно-рецессивно. Причем у гетерозигот кровотечения бывают незначительны; у гомозигот с дефицитом ФХI осложнений, связанных с кровоточивостью, отмечается не много. Но при травме или хирургическом вмешательстве не исключено возникновение сильного кровотечения с формированием гемартроза и гематомы. При лабораторной диагностике выявляются следующие признаки: увеличение АПТВ, ПВ и ТВ в норме; нормальные показатели сосудисто-тромбоцитарного гемостаза; - уровень ФХI в плазме снижен или равен нулю. Парагемофилия (дефицит ФV) наследуется аутосомно-доминантно или аутосомно-рецессивно. Для заболевания характерен геморрагический синдром, выраженность которого зависит от степени дефицита в плазме ФV. Наиболее тяжелая кровоточивость наблюдается у больных, у которых уровень ФV менее 2%. При средней тяжести он составляет 2 – 6%, при легкой – 6 – 16%. При данном заболевании отмечаются петехии, экхимозы, кровоподтёки, носовые, десневые, желудочно-кишечные кровотечения, меноррагии. У больных при выраженных формах заболевания часты длительные кровотечения после удаления зубов, при травмах, порезах. При лабораторной диагностике выявляются: увеличение АПТВ и ПВ; нормальные показатели сосудисто-тромбоцитарного гемостаза; - снижение или полное отсутствие в плазме уровня ФV. Приобретенная коагулопатия (диссеминированное внутрисосудистое свертывание, синдром дефибринации, ДВС-синдром) ДВС-синдром – неспецифический общепатологический процесс, характеризующийся генерализованной активацией системы гемостаза-антигемостаза, при котором происходит рассогласование систем регуляции агрегатного состояния крови. Этиологическим фактором заболевания являются: генерализованные инфекции, септические состояния; шок любого происхождения; обширные хирургические вмешательства, в том числе акушерская патология (разрыв плаценты, эмболия околоплодными водами, криминальный аборт); злокачественные опухоли; обширные повреждения тканей, тканевая эмболия, ожоги; иммунные, аллергические и иммунокомплексные болезни; массивные кровопотери, трансфузии; отравления гемокоагулирующими змеиными ядами, химическими и растительными веществами, внутрисосудистый гемолиз любого происхождения; – острые гипоксии, гипотермия, гипертермия с дегидратацией. В основе патогенеза ДВС-синдрома лежат следующие механизмы: системное поражение и неполноценность сосудистого эндотелия; активация свертывающей системы и тромбоцитов; первичная или вторичная депрессия противосвертывающей системы (антикоагулянтной – дефицит антитромбина III и фибринолитической – резкое повышение антиплазминовой активности, дефицит плазминогена и его активаторов). Таким образом, при ДВС-синдроме нарушается как сосудистотромбоцитарный, так и коагуляционный виды гемостаза. Основными звеньями патогенеза тромботического синдрома являются: повреждение тканей, которое сопровождается поступлением в кровоток огромного количества прокоагулянтов (тканевого тромбопластина) и генерализованной активацией факторов свертывающей системы крови с преобладанием внешнего механизма свертывания; системное поражение сосудистого эндотелия, которое может быть обусловлено действием бактерий (менингококки), эндотоксинов, вирусов; оно сопровождается выделением эндотелиальных прокоагулянтных факторов, активацией тромбоцитов и гиперактивацией внутреннего механизма свертывания крови, нарастающего потребления и, как следствие, дефицита факторов противосвертывающей системы (антитромбина III, протеинов С и S и др.); стимуляция тромбоцитов и макрофагов, когда при бактериальных, вирусных инфекциях, действии иммунных комплексов, эндотоксинов происходит прямая или опосредованная (через активацию макрофагов и выделение цитокинов) активация тромбоцитов, которая сопровождается формированием внутрисосудистых тромбоцитарных микроагрегатов (тромбов); следствием этого являются тромбоцитопения потребления и нарастающая капилляро-трофическая недостаточность. Для патогенеза геморрагического синдрома характерны: гиперактивация механизмов коагуляции крови, сопровождающаяся нарастающим потреблением факторов свертывающей системы и тромбоцитов, что приводит к тотальному дефициту факторов свертывания (коагулопатия и тромбоцитопения потребления) и к развитию геморрагического синдрома; образование тромбина в сосудистом русле, сопровождающееся значительной активацией фибринолитической системы, которая также играет важную роль в развитии практически не останавливающихся кровотечений. В развитии ДВС-синдрома по гемостазиологической характеристике выделяют следующие стадии: 1) гиперкоагуляция и агрегация тромбоцитов; 2) переходная; 3)гипокоагуляция (коагулопатия потребления); 4) восстановительная. В клинической картине ДВС-синдрома отмечаются: в 1-й стадии – симптомы основного заболевания и признаки тромбогеморрагического синдрома (с преобладанием проявлений генерализованного тромбоза); гиповолемия, нарушение микроциркуляции, дисфункция и дистрофические изменения в органах, нарушения метаболизма; во 2-й стадии появляются признаки полиорганного повреждения и блокады системы микроциркуляции паренхиматозных органов, геморрагический синдром (петехиально-пурпурный тип кровоточивости); в 3-й стадии к указанным нарушениям присоединяются признаки полиорганной недостаточности (острая дыхательная, сердечно-сосудистая, печёночная, почечная, парез кишечника) и метаболические нарушения (гипокалиемия, гипопротеинемия, метаболический ацидоз, алкалоз), а также анемический синдром, геморрагический синдром по смешанному типу (петехии, гематомы, кровоточивость из слизистых оболочек, массивные желудочно-кишечные, лёгочные, внутричерепные и другие кровотечения, кровоизлияния в жизненно важные органы); в 4-й стадии (при благоприятном исходе) основные витальные функции и показатели гемостаза постепенно нормализуются. ДВС-синдром может протекать: молниеносно (от нескольких минут до нескольких часов или одних суток); – остро (1-10 суток); подостро (до 1 месяца); хронически (более 1 месяца); рецидивирующе (волнообразно).     |
