Курсовая. Курсовая работа По дисциплине Физикохимические методы анализа На тему Физикохимические методы исследования качества воды
 Скачать 498.33 Kb. Скачать 498.33 Kb.
|

1 2 МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ ДОНЕЦКОЙ НАРОДНОЙ РЕСПУБЛИКИ ГОСУДАРСТВЕННОЕ ПРОФЕССИОНАЛЬНОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ «ДОНЕЦКИЙ ТЕХНИКУМ ХИМИЧЕСКИХ ТЕХНОЛОГИЙ И ФАРМАЦИИ» Курсовая работа По дисциплине « Физико-химические методы анализа» На тему «Физико-химические методы исследования качества воды» Студентки III курса ,группы 9АК-19 Очной формы обучения Верхотуровой Дарьи Николаевны Научный руководитель Марченко И.В Донецк-2021 Содержание Вступление 1. Требовaния, предъявляемые к питьевой воде 1.1Гигиенические требования к кaчеству питьевой воды .2 Производственный контроль качества питьевой воды .3 Органолептические показатели качества питьевой воды .4 Показатели радиационной безoпасности питьевой воды .5Показатели физиологической полноценности качества воды 2. Метод атомно-абсорбционной спектроскопии .1 Эмиссионная фотометрия пламени .2 Иoнометрия. Oсобенности выбора и эксплуатации электродов 3. Физико-химические методы исследования качества воды 3.1 Определение железа, меди и цинка в природных водах методом атомно-абсорбционной спектроскопии .2 Определение ионов калия и натрия в речной или водопроводной воде методом добавок (эмиссионная фотометрия пламени) .3 Oпределение фторид-иона в питьевой воде с помощью фторид-селективного электрода Вывод Список использованной литературы ВСТУПЛЕНИЕ в 1913 М.С. Курнаков, обобщив количественные методы химических исследований, использованных Ломоносовым, Лавуазье, Дальтоном, Менделеевым, Гиббсом, Вант-Гоффа, Розебома, Ле Шателье, Схрейнемакерсом и многими другими учеными, предложил метод исследования веществ, «общий прием которого состоит в количественно изучении свойств равновесных систем , образованных двумя и более компонентами, в зависимости от их состава »назвать физико-химическим анализом . Инструментальные или физико химические методы анализа основаны на измерении с помощью приборов определенных физических свойств системы, является функцией количества компонента, который определяют, в пробе, анализируют. Инструментальные методы анализа имеют ряд преимуществ по сравнению с классическими методами: значительно более высокую чувствительность, селективность, экспрессность, объективность, возможность автоматизации и компьютеризации процесса анализа. В производственных условиях большое значение имеет скорость выполнения анализа - экспрессность анализа. Физико-химические методы анализа отличаются большой избирательностью, чувствительностью, скоростью выполнения аналитических операций. В физико-химических методах анализа определяют изменения физических свойств системы (коэффициента преломления света, поглощение света, электропроводности), которые происходят в результате химических или электрохимических реакций. При выполнении анализов физико-химическими методами точку эквивалентности (момент окончания реакции) определяют не визуально, а с помощью приборов, фиксирующих изменения тех или иных свойств исследуемого вещества. В данной курсовой работе изложены теоретические основы некоторых оптических методов анализа, рассмотрены методики проведения определений и аппаратуру, используется в данных методах. Актуальность темы. Человеческий организм примерно на 75% состоит из воды. Считается, что мозг состоит из воды на 85% и отличается исключительной чувствительностью к обезвоживанию. Мозг постоянно омывается соленой спинномозговой жидкостью. Присутствует во всех клетках и тканях, играя главную роль во всех биологических процессах от пищеварения до кровообращения, вода выполняет много важных функций. Наше тело постоянно нуждается в пополнении запаса чистой водой. Вода должна стать для нас самым ключевым ингредиентом, если мы хотим иметь здоровое тело и отличное самочувствие. Ничто так не влияет на наше здоровье, как потребление воды. Поэтому, тема определения качества воды всегда будет актуальной. 1. Требования, предъявляемые к питьевой воде ДСанПиН №383 Об утверждении Государственных санитарных правил и норм "Вода питьевая. Гигиенические требования к качеству воды централизованного хозяйственно-питьевого водоснабжения" 28.10.09 1.1 Гигиенические требования к качеству питьевой воды химическое исследование качество вода Гигиенические требования, определят пригодность воды для питьевых целей, включают: безопасность в эпидемическом отношении; безвредность химического состава; благоприятные органолептические свойства; радиационной безопасности. Качество питьевой воды зависит от ее состава и свойств: в водоисточников; при поступлений в водопроводную сеть; в точках водоразбора. Безопасность питьевой воды в эпидемическом отношении определяется показателями, характеризующими с достаточно высокой вероятностью отсутствие в ней опасных для здоровья потребителей бактерий, вирусов и других биологических включений. Примечания: * - для 95% проб воды в водоснабжающей сети, исследуются в течение года; ** - для 98% проб воды, поступающей в водоснабжающую сеть, и исследуются в течение года; при превышении индекса БГКП на этапе идентификации колоний, выросших дополнительно проводят исследование на наличие фекальных колиформ; *** - при обнаружении фекальных колиформ в 2-х последовательно отобранных пробах воды следует начать в течение 12:00 исследования воды на наличие возбудителей инфекционных заболеваний бактериальной или вирусной этиологии (по эпидситуации). По паразитарным показателям (клетки, цисты: лямблий, криптоспоридий, а также в случае эпидосложнений - дизентерийная амеба, балантидий, хламидий и др .; клетки, личинки, яйца гельминтов) питьевая вода должна соответствовать требованиям, приведенным в табл. Паразитологические показатели безопасности питьевой воды 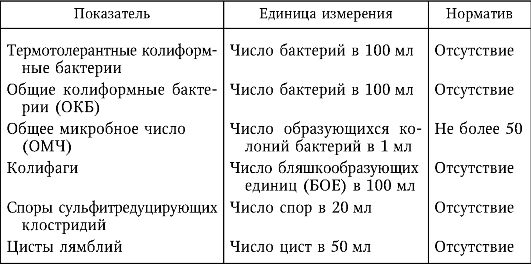 Безвредность химического состава питьевой воды определяется показателями, которые с достаточно высокой вероятностью характеризуют отсутствие в ней опасных для здоровья веществ (компонентов), встречающихся в природных водах, появляются в воде вследствие загрязнения водоисточников или в процессе водообработки в концентрациях, предельно допустимые величины которых установлены результатам санитарно-токсикологических исследований. По токсикологическим показателям питьевая вода должна соответствовать требованиям, приведенным в табл.. Токсикологические показатели безвредности химического состава питьевой воды  Вода не должна содержать другие токсичные компоненты (ртуть, таллий, кадмий, нитриты, цианиды, хром (+6), 1,1-дихлоретилен, 1,2-дихлорэтан, бенз (а) пирен) в концентрациях, определяемых стандартными методами исследований . При проведении обеззараживания воды концентрация остаточных количеств дезинфектантов, которые определяются не реже чем раз в час, должен составлять: при обеззараживании питьевой воды хлором содержание остаточного свободного хлора в воде на выходе из резервуаров чистой воды должно быть 0.3-0.5 мг / л (если продолжительность контакта хлора с водой не менее 30 мин.), а содержание остаточного связанного хлора - 0.8 -1.2 мг / л (если продолжительность контакта хлора с водой не менее 60 мин.). При совместной наличии в воде свободного и связанного хлора разрешается осуществление контроля за одним из них: по остаточному свободным хлором (при его концентрации более 0.3 мг / л) или по остаточному связанным хлором (при концентрации остаточного свободного хлора меньше 0.3 мг / л); при обеззараживании воды озоном концентрация остаточного озона на выходе из камеры смешения должно быть 0.1-0.3 мг / л при продолжительности контакта не менее 4 мин. Взаимосвязь дозы дезинфектанта (C, мг / л) и времени (T, мин), необходимого и достаточного для обеспечения эпидемической безопасности обрабатываемой воды во время ее прохождения к первому потребителю, определяет критерий "CxT", который может быть установлен экспериментально для каждого конкретного воды с учетом показателей ее хлорпоглинальности. При использовании в процессе водоподготовки коагулянтов, дезинфектантов или иных реагентов, разрешенных Министерством здравоохранения Украины для применения в практике хозяйственно-питьевого водоснабжения, их остаточные количества в воде не должны превышать соответствующих нормативных значений. 1.2 Производственный контроль качества питьевой воды Производственный контроль качества питьевой воды включает в себя: - определение состава и свойств воды источника водоснабжения и питьевой воды в местах водозабора, перед поступлением ее в водопроводную сеть, распределительной сети; - входной контроль наличия сопроводительной документации (технических условий, сертификата соответствия или гигиенического сертификата (гигиенического заключения) на реагенты, материалы и другую продукцию, используемые в процессе водоподготовки; - входной выборочный контроль продукции, используемой в процессе водоподготовки, на соответствие требованиям нормативной документации на конкретный продукт; - в соответствии с технологическим регламентом пооперационный контроль оптимальных доз реагентов, вводимых для очистки воды; разработку графика контроля, согласованного с территориальными органами Госсанэпиднадзора России и (или) ведомственного санитарно-эпидемиологического надзора в установленном порядке, который должен содержать контролируемые показатели; периодичность и количество отбираемых проб; точки и даты отбора проб и т. д.; - экстренное информирование центров санэпиднадзора обо всех случаях результатов контроля качества питьевой воды, не соответствующих гигиеническим нормативам, прежде всего превышения по микробиологическим и токсикологическим показателям; - ежемесячное информирование центров санэпиднадзора о результатах производственного контроля. 1.3 Органолептические показатели качества питьевой воды Потребительские свойства воды напрямую определяются ее органолептическими показателями. Исследование органолептических свойств – это первая ступень контроля качества воды, которую никак нельзя пропустить. При этом некоторые из них (запах и привкус) нельзя измерить, они подвергаются экспертной оценке. Благоприятные органолептические свойства питьевой воды определяются совокупностью значений, регламентируемых органолептическим показателям качества и физико-химическими характеристиками воды (по содержанию в воде компонентов, которые влияют на органолептические показатели). Органолептические показатели и предельно допустимые концентрации компонентов, которые нормируются по их влиянию на органолептические свойства питьевой воды. Вода не должна содержать другие компоненты, способные изменять ее органолептические свойства, - цинк, поверхностно-активные вещества, нефтепродукты, фенолы в концентрациях, определяемых стандартными методами исследований. 1.4 Показатели радиационной безопасности питьевой воды Радиационная безопасность питьевой воды определяется по предельно допустимыми уровнями суммарной объемной активности альфа- и бета-излучателей (природных). В случае превышений уровней следует провести изучение радионуклидного состава исследуемых проб воды на предмет его соответствия нормам радиационной безопасности. 1.5 Показатели физиологической полноценности качества воды Показатели физиологической полноценности питьевой воды визначаютьадекватнисть ее минерального состава биологическим потребностям организма. Они основаны на целесообразности для ряда биогенных элементов учета не только максимально допустимых, но и минимально необходимых уровней их содержания в воде. 2.Метод атомно-абсорбционной спектроскопии В этом методе мы рассмотрим поглощения атомами, а не молекулярными соединениями. Чтобы созерцать оптические свойства свободных атомов, необходимо пробу перевести в газообразное состояние, а это в большинстве случаев требует испарения жидкости или твердого вещества и последующей диссоциации молекул на свободные атомы. Атомизация Существует много способов атомизации соединений металлов, осуществляемых в большинстве случаев за счет тепловой энергии электричества или пламени. Для оптимального перехода в атомный пар необходим строгий контроль за температурой. Слишком высокая температура может быть так же неблагоприятна, как и слишком низкая, так как часть атомов ионизируется и, следуя из этого, поглощает при ожидаемых длинах волн. Но, с другой стороны, высокая температура способствует снижению влияния матрицы, поэтому нужно найти компромисс между этими температурами. Атомизаця пламенем. На рис. 2.1 изображена горелка, которая используется в атомно-абсорбционной спектроскопии пламенем (ААСП).  Горючий газ и газ-окислитель подаются в смесительную камеру, где они проходят через ряд перегородок, обеспечивающих их полное смешение, и попадают в верхнюю часть горелки. Отверстие горелки имеет форму длинной узкой щели, позволяет получить пламя в виде узкой полосы. Раствор, который анализируется засасывается в смесительную камеру с помощью небольшой воздушной форсунки. При использовании такого распылителя получают капли различного размера, что может быть причиной плохого воспроизведения. При прохождении через перегородки смесителя большие капли задерживаются, так что в пламя попадают небольшие однородные по размеру капли. Горелка с предварительным смешиванием газов не совсем безопасен в работе, потому что, если пламя попадет в смесительную камеру, состоится сильный взрыв. Для того, чтобы свести к минимуму вероятность проскока пламени в камеру, щель горелки нужно сделать как можно более узкой (для того, чтобы газы продувались в ней с большой скоростью), а металлический ободок вокруг щели можно массивным, так чтобы тепло легко отводилось. Но даже в этом случае, если не регулировать газовый поток должным образом, взрыв возможен. В горелках, которые продаются продуманные меры безопасности при проскок пламени в камеру. При эксплуатации горелки необходимо всегда строго соблюдать правила техники безопасности. В качестве окисляющего и горючего газов в ААСП чаще всего выбирают сжатый воздух и ацетилен. Максимальная температура, которую достигают составляет примерно 2200 ° С. Если нужна более высокая температура, воздух можно заменить оксидом азота (N2O), который распадается с образованием смеси азота и кислорода в соотношении 2: 1, тогда как для сжатого воздуха это соотношение равно 4: 1; максимальная температура, которую можно получить при горении ацетилена, составляет почти 3000 ° С. В горелках с предварительным смешиванием газов нельзя использовать чистый кислород, так как пламя распространяется так быстро, что проскок в камеру неизбежен. Пламя - удобное и воспроизводительное источник тепла, но в качестве рабочей кюветы этот источник далеко от идеала, потому что два эндотермических процессы (испарение растворителя и последующая атомизация) должны пройти за очень короткий промежуток времени, каким частицам удается пролететь сквозь пламя, а не атомизуючись. Кроме того пламя вносит значительные случайные флуктуации в эффективную длину оптического пути вследствие турбулентности, а это приводит к лишнего шума при получении сигнала. Некоторого улучшения можно добиться, используя пламя только как источник тепла. Один из способов заключается в том, что растворенную пробу помищяють в небольшой металлический челнок или чашечку, которая называется чашкой Дельво , и высушивают на горячей пластинке. Чашечку после этого закрепляют над пламенем для испарения пробы. Пар, образовавшийся попадает через центральное отверстие в горизонтальную кварцевую трубку , которая нагревается узким пламенем, где собственно и испускаемого излучения, которое измеряют. Кварцевая трубка играет роль сдерживающего капилляра, с помощью которого увеличивается время наблюдения за атомами. Это способ оказался полезным при обнаружении следов свинца. Обычно используются никелевые чашечки, но при анализе кислых проб никель заменяют танталом или другим инертным материалом.  НЕ пламенные атомизаторы. За последнее тридцатилетие вместо пламени все более широко используют электронагрева. Было предложено несколько типов нагревателей, из которых наиболее удачным оказался устройство, которое состоит из небольшой графитный трубки [3], которая нагревается при пропускании через нее тока большой силы (до 500 А) при низком напряжении. Внутренний диаметр графитный трубки (модели Вариан) составляет несколько миллиметров, а длина около сантиметра. Сверху трубки имеется небольшое отверстие, через который вводится растворена проба.  Атомизатор угловыми электродами фирмы Varian модели CRA-90. Графитовая кювета представляет собой небольшой цилиндр, закрепленный в поперечном положении между двумя графитовыми стержнями, являются и механическими держателями, и проводниками тока. С помощью шланга слева подводится инертный газ. На фотографии показано введение пробы пипеткой (Varian Techtron). Внутренняя поверхность покрыта устойчивой формой углерода, которая называется пиролитическим графитом. Подобные трубки изготавливаются также другими фирмами и отличаются деталями. Трубка, которая часто называется графитовой кюветой, должна находиться в атмосфере инертного газа, например аргона, для предотвращения окисления пробы и горючего углерода. При работе с жидкими пробами обычно приходят к программированного повышения температуры графитовой кюветы в три этапа. Сначала ее поднимают примерно до 300 ° С и поддерживают в течение минуты для испарения растворителя. Затем поднимают температуру до 1700 ° С и выдерживают еще около минуты; при этом сгорают органические вещества. Только после этого температуру повышают до уровня необходимого до уровня диссоциации неорганического соединения на атомы; иногда нужно поднять ее до 3000 ° С. Конструкция большинства приборов позволяет оператору менять интервал времени и температуру в зависимости от характера пробы. В процессе сушки на ленте самописца с разверткой по времени часто оказывается посторонний пик, связанный с поглощением паров растворителя. На стадии обугливания при наличии большого количества органических веществ также возможно проявление пика. Аналитическую информацию получают, измеряя высоту пика, который наблюдается при конечной температуре. На высоту пика влияют скорость нагрева и размеры кюветы [4], поэтому при сравнении результатов измерения неизвестной пробы результатам измерения стандартов нужно точно воспроизводить эти и другие параметры, которые изменяются. Летучие гидриды. Для элементов, которые образуют летучие гидриды, а именно As, Bi, Sb, Se и Te , существует другой способ подготовки пробы. Гидриды получают с помощью реакции солей этих элементов с щелочным раствором боргидрид натрия. Летучие вещества затем устраняются из раствора с потоком воздуха прямо в атомизатор атомно-абсорбционного спектрофотометра. Часто используется водородно-воздушная пламени; кварцевая трубка-кювета, которая подогревается извне позволяет спуститься к более низких концентраций вследствие увеличения времени пребывания в ней атомов. Этим способом определяли также олово и свинец, но результаты оказались менее удовлетворительными. Аналогичным путем можно восстановить ртуть в элементного состояния (если она была в ишому степени окисления) и перенести ее с потоком воздуха в кварцевую трубку-кювета, которая не требует нагрева [6].источник излучения Почти во всех атомно-абсорбционных спектрометрах используются лампы, которые дают линейчатый спектры, характерные для отдельных элементов. Эти лампы вместе с обычными монохроматорах средней разрешающей силы гораздо эффективнее источников непрерывного света. Монохроматор служит для выделения нужной линии испускания, а не для сужения полосы поглощения. Нецелесообразности использования источников непрерывного излучения связана с тем, что линии поглощения нейтральных атомов в пламени или кювете чрезвычайно узкие. Ширина их составляет около 0,001нм, тогда как полуширина полосы пропускания обычного монохроматора несколько десятых нанометра. Сравнение излучения источника непрерывного спектра и поглощения атомного пару. Кривая а представляет собой полосу длин волн, прошедших через монохроматор; кривая бы, намного более узкая кривой а, - поглощение атомами в пламени .Площадь под кривой а, которая соответствует пропущенном излучению источника, испытывает лишь очень незначительных уменьшений за счет линии атомной абсорбции бы.Наибольшим успехом в качестве источника линейчатого спектра в ААС пользуется лампа с полым катодом. Она представляет собой стеклянный или кварцевый баллон, в котором расположены два электрода. Один из них (катод), который имеет чашеобразную форму, изготовлен из какого-либо определенного элемента . Материал анода не имеет значения. Лампа заполнена благородным газом под низким давлением. При подаче напряжения 100 - 200 В после непродолжительного разогрева возникает тлеющий разряд с участием большинства частиц, которые попадают из полого катода. Положительные ионы инертных газов бомбардируют катод, выбивая атомы металла; этот процесс называется распылением. Атомы поглощают энергию и возбуждаются, выпуская характеристическое излучение. Оно состоит из дискретных линий этого металла и линий газа-наполнителя, который выбирают таким образом, чтобы спектральные помехи от него при определении данного металла были минимальными. Излучения лампы с полым катодом, который изготовлен из никеля. Атомы никеля в пламени в заметной степени поглощают только излучение при 232,0 нм . Надо обратить внимание на то, что чувственная линия при 232 нм окружена многими другими линиями никеля, поглощение которых паром никеля нежелательно. Нужную линию можно изолировать с помощью монохроматора с узкой полосой пропускания. На рис. сравнивается спектр излучения лампы с полым катодом (а) с тем же спектром после прохождения излучения через монохроматор (б) и показано влияние поглощения паром металла (в).  Рис. 2.7. Атомная абсорбция: а - спектр излучения лампы с цинковым катодом (ширина линии 213,9 нм увеличена для наглядности) б - лишние линии цинка отсекаются монохроматором, который настроен на 213,9 нм; в - интенсивность линии при 213,9 нм резко уменьшается в результате поглощения атомами цинка (Unicam Instruments, Ltd.). Полоса поглощения всегда шире эмиссионной линии, хотя и узкий полосы пропускания монохроматора. Очевидно, что уменьшение интенсивности потока излучения, которое прошло в детектор, напрямую зависит от числа атомов металлов в зоне нахождения пробы. Можно изготовить лампы с полым катодом, в которых чаша покрыта смесью нескольких металлов (при условии, что они не мешают друг другу при спектральном определении и для их испарения необходимо примерно одинакова энергия). Это делает возможным определение нескольких элементов без замены лампы. В качестве примера можно назвать лампы, катод которых состоит из Ca, Mg, и Al; Fe, Cu и Mn; Cu, Zn, Pb и Sn; Cr, Co, Cu, Fe, Mn и Ni. Лампе с полым катодом, как и многим другим источникам, для выхода на постоянный режим работы потребуется некоторое время после включения. Особенно это удобно при определении с помощью одноэлементных ламп нескольких элементов в одной пробе. Один из путей преодоления этой сложности заключается в использовании турели, на которой закреплено несколько ламп, которые находятся в рабочем режиме, так что любую из них можно установить в нужное положение поворотом турели. Для уменьшения задержки при смене ламп используют двухлучевой систему корректировки яркости лампы. Источником в ААС может служить и безэлектродная разрядная лампа, которая представляет собой запаянную кварцевую трубку, которая содержит небольшое количество чистого металла под низким давлением инертного газа. Возбуждение возникает под действием микроволнового поля волнового резонатора, причем выпускается, по сути, тот же спектр, и лампой с полым катодом. Можно также пользоваться источником непрерывного спектра в сочетании с монохроматором высокого разрешения. ОьХевер и соавторы [6-7] показали, что ксеноновая лампа большой мощности с эшелетт в качестве монохроматора позволяет сузить полосу поглощения, чтобы получить правильное значение оптической плотности. Достоинством такой ламы является то, что при переходе от одного элемента к другому достаточно просто изменить длину волны. Авторы описали прибор с 16 фото-умножителем, на котором возможно одновременное определение такого же количества элементов, но с меньшей чувствительностью, чем с помощью лампы с полым катодом. В атомно-абсорбционных спектрофотометрах заводского изготовления непрерывные источники не используются. Поправка на поглощение фона В ААС в отличие от спектрофотометрии растворов нельзя учесть влияние фона с помощью простой двухлучевой системы. Такой прием требовал бы дубликатов пламени или графитовых кювет (один рабочий, другой сравнения), а сделать их оптически равноценными было бы чрезвычайно трудно. Тем не менее, при качественных измерениях поправка на фон является весомой.  Рис. 2.8. Основные узлы АА-спектрофотометра: а - схема без прерывания излучения (выпуска нагретой пробы сливается с излучением лампы с полым катодом, приводит к погрешности) б - схема с прерыванием излучения (детектор фокусирует только излучение лампы). Фоновый сигнал частично обусловлен излучением, выделяется собственно нагретой пробоя. Это источник фонового излучения, характерное только для AAC, обусловлено неизбежным возбуждения атомов анализируемой вещества, самовольно выпускающих фотоны при тех же длинах волн, при которых изучается поглощения. Как схематично показано на рис. 2.8, а, если не применить меры безопасности, интенсивность света, наблюдается Рспост, будет состоять из Р0Т и Ре, где Р0 - интенсивность падающего излучения, Т - пропускание пробы и Ре и судьба интенсивности выпуска пробы, попадает в детектор. Излучения с интенсивностями Ро и Ре имеют одинаковую длину волны, и поэтому при попадании (через монохроматор) в детектор не отличаются друг от друга. Устранить это можно с помощью прерывателя, который прерывает излучение от лампы с полым катодом, как показано на рис. 2.8, б, не влияя на излучение из пробы. Для того, чтобы сигнал, обусловленный выпуском пробы, вичитався из общего сигнала, можно поставить электронный усилитель, работающий синхронно с прерывателем. В некоторых устройствах такой результат получают при возбуждении лампы с полым катодом электрическими импульсами; при этом вместо постоянного излучения, прерывается с помощью диска прерывателя, получается переривчате излучения. Вклад в фон вносит также поглощение других компонентов пробы. Описано несколько способов снижения такого рода препятствий. Первый из них основан на использовании одновременно источника непрерывного излучения, например водной или дейтериевой лампы и источники линейного спектра [8-9], как показано на рис.2.9.  Рис.2.9. АА-спектрофотометр с водородной (или дейтериевой) лампой, с помощью которой вводится поправка на фон. Излучения вспомогательной лампы проходит через пробу вместе с резонансным излучением лампы с полым катодом. Электронная система сортирует сигналы от обоих источников и дает их отношения. В одних устройствах (фирмы Perkin-Elmer) оба сигнала видоизменяют с помощью вращающего зеркала, в других (фирмы Instrumentation Laboratories) через оптическую систему проходят сразу оба потока, прерываемых с разной скоростью. Оба потока равномерно ослабляются за счет поглощения фоном или рассеяния, но пробой заметно поглощается только резонансное излучение. Второй метод устранения фонового сигнала основан на эффекте Зеемана. И выпуска и поглощения УФ и видимого излучения связано со свойствами электронов, вращающихся в атомах, поэтому неудивительно, что эти явления сильно зависят от наличия магнитного поля. Теоретически предусмотрено и практически доказано, что если источник излучения (лампу с полым катодом) или поглощающую пробу поместить в поперечное магнитное поле, каждая линия выпускающего излучения расщепляется в простейшем случае на три линии, одна из которых имеет несколько большую длину волны, вторая немного меньше, то есть, если на оптическом пути поставить поляризатор, то их можно отличить.  Рис. 2.10. Оптическая схема АА-спектрофотометра фирмы Hitachi (модель 180/70), на которой показано направление магнитного поля Зеемана вокруг графитового атомизатора (Hitachi, Ltd). Вокруг атомизатора (графитовая кювета, как на рисунке, или пламя) размещают постоянный магнит. Между атомизатором и лампой с полым катодом помещают поляризатор, который можно повернуть так, чтобы плоскость поляризации была параллельна или перпендикулярна направлению магнитного поля. При поляризации излучения под прямым углом к полю не поглощается атомным паром, а при параллельной поляризации поглощается, как магнитного поля и не было. Однако поглощение, связанное с фоном, не меняется ни в том, ни в другом случае, поэтому, отнимая друг (при перпендикулярной поляризации) от другого (при параллельной поляризации), получают спектр поглощения с поправкой на фон. Преимущество этого способа над вышеописанным заключается в том, что здесь используется только одна лампа, поэтому не возникает проблем, связанных с выравниванием потоков. В спектрофотометр с поправкой, что вводится на основе эффекта Зеемана, возможно и другое расположение компонентов . Их относительные достоинства и недостатки обсуждаются в обзоре . Третий способ учета фона основан на модулировании длины волны. Харнлы и ОьХевер [14] вывели выражение для оптической плотности пробы для применения которого нужно выделить постоянную и переменную составляющую тока фото умножителем. Переменную составляющую находят по сдвига синусоиды длин волн при колебании кварцевой пластинки, помещенной в центре выходной щели: = Lg (P0 / P) = lg [(DC + 0,38AC) / (DC-0,62AC)] Где AC и DC - полный размах колебаний переменной и постоянной составляющих соответственно. Авторы показали, что этот способ почти вдвое эффективнее способов с использованием дейтериевой лампы или эффекта Зеемана. Впоследствии появилось сообщение о еще одном способе введения поправки на фон , достаточно интересное, чтобы рекомендовать его По этому способу поправку получают с помощью самой лампы с полым катодом. Сначала, чтобы измерить суммарную оптическую плотность элемента и фона, в лампу подают импульс тока малой силы (12 мкА). Затем, чтобы расширить эмиссионную линию лампы, подают короткий импульс тока большей силы (250 мкА). Относительное поглощением элементом пробы резко уменьшается, тогда как фоном поглощается постоянная часть излучения. Разница между двумя сигналами предоставляет истинную аналитическую информацию. предел обнаружения Чувствительность АА-методов связана сложной зависимостью с оптическими свойствами атомного пару, температурой, относительной шириной линии лампы и поглощающих частиц и характером оптической системы. В целом предела обнаружения при использовании неполумьянои ААС в 100 и более раз ниже, чем пламенной, хотя и присутствуют некоторые исключения; так, например, границы вивлення K, Fe, Sn тем и другим методами одинаковы. препятствия В пламенной ААС причиной епершкод могут быть химические реакции в пламени. Основные трудности связаны с неполной диссоциации или с образованием трудно летучих соединений. Некоторые элементы, например Те, Al, i V, окисляются в пламени, образуя соединения, устойчивые при температуре воздушно-ацетиленовой пламени; большинство таких соединений разлагаются в N2O-ацетиленовом пламени. В других случаях элемент образует устойчивые соединения с некоторыми другими компонентами пробы. Например, рассмотрим определение Sr в присутствии Al или Si. На рис. 2.11 показано, как сильно они мешают анализа.  Рис. 2.11. Один из способов устранения химических помех в ААС. Добавление соли лантана защищает стронций от мешающих действия по стороны алюминия и кремния. Эти препятствия можно почти полностью устранить добавлением к пробе небольшого количества соли лантана, связывающей Al или Si и не взаимодействует с Sr. Препятствия возможны и при определении солей Са. Соединения кальция полностью диссоциируют в N2O-ацетиленовом пламени, но температура его настолько высока, что заметная часть атомов Са ионизируется, и как следствие, не попадает в число определяемых атомов. Это можно регулировать добавлением более легко ионизирующего элемента, например Na; при этом атомы Са остаются в нейтральном состоянии. Вторым источником помех является изменение вязкости или других свойств растворов, влияющих на легкость распыления и переноса пламени. В результате для двух растворов с одинаковой концентрацией металла, но с разным количеством чужеродных веществ, можно получить различные показания прибора. Те же проблемы возникают при работе с графитовым атомизатором. Только здесь наиболее вероятно образование соединений с углеродом, а не с кислородом. Карбиды алюминия и кремния при нагревании диссоциируют, но карбида вольфрама и бора более устойчивы при нагревании. 2.1 Применение атомной абсорбции Атомная абсорбция используется для обнаружения большого количества металлов, особенно в следовых количествах. Она широко применяется в таких областях, как анализ воды и фармацевтических препаратов, а также в металлургии. При любом обнаружении совершенно необходимо скрупулезно соблюдать экспериментальных условиях: нужно найти специальные рекомендации и строго их соблюдать, а если их нет, то провести методические исследования. Устройства, выпускаемые промышленностью, содержат обширные инструкции, содержащие методики выявления всех известных металлов в различных матрицах. Монография Ван Луна [5] является прекрасной сборкой методик и содержит обсуждение принципов АА-анализа. 2.2 Эмиссионная фотометрия пламени Метод основан на измерении интенсивности излучения атомов и молекул, которые возбуждаются в пламени. Пламя образуется при сгорании различных органических веществ в окислителях (табл.2.1.). Температура пламени не высока (1700 - 3000 ° С), однако достаточна для возбуждения резонансных линий атомов, которые наиболее легко возбуждаются (менее 600 кДж / моль). Пламя состоит из нескольких зон. В спокойном (ламинарном) пламени можно выделить внутренний и внешний конусы, а также промежуточную зону Рис. 2.12. Температура (° С) в пламени: а - смесь светильного газа с воздухом; б - ацетилен с кислородом. Во внутреннем (восстановительном) конусе горючие газы окисляются не полностью и в спектре этой части пламени видны полосы молекул OH, C2 и других. В верхнем (окислительной) конусе топливо сгорает полностью в CO2 и H2O. Температура отдельных частей пламя зависит от состава смеси, горит. Для целей анализа обычно используют верхнюю часть пламени, где собственное излучение пламени, обусловлено продуктами сгорания (фон), меньше всего. Пламя - удобный источник возбуждения, поскольку спектр веществ, которые возбудились в пламени, представлен в основном резонансными линиями, достоинствами пламя являются также его стабильность (при стабильном давлении горючих газов), простота конструкций горелок, малую степень ионизации атомов. Однако пламя имеет такие недостатки, как наличие фона и сравнительно низкие температуры, при которых число атомов, которые возбуждаются небольшой (например, при 2200 ° С всего 0,02% атомов натрия находятся в возбужденном состоянии). Процессы в пламени Вещество, которое исследуется в пламени, обычно вводят в виде растворов (распыляют), для анализа можно внести в пламя и твердую пробу. При этом в пламени протекает ряд процессов: испарения растворителя с образованием твердых частиц вещества, испарение твердых частиц с образованием атомного пара, диссоциация молекул на атомы, частичная ионизация, возбуждение атомов, возвращение атомов в исходное состояние с выделением квантов света (рис. 2.13 ).  Рис. 2.13. Процессы, происходящие с веществом MX в пламени. Интенсивность излучения атомами (молекулами) пропорциональна их концентрации в пламени, которая в свою очередь пропорциональна концентрации ионов в растворе. Эта прямолинейная зависимость соблюдается при постоянном коэффициенте k, на значение которого могут влиять такие препятствия, как самопоглощение, ионизация, образование сложно летучих соединений, изменение режима работы и др. Интенсивность излучения в эмиссионных методах измеряют в пламенных фотометрах и спектрофотометрах, превращая световой поток в электрический ток (фототок) с помощью фотоэлементов. Пламенные фотометры Схема пламенного фотометра (рис. 2.14) включает: распылитель раствора; горелка, в который подается горючая смесь; монохроматор; фотоэлемент и регистрирующее устройство. 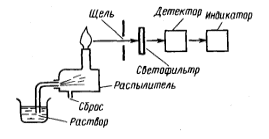 Рис.2.14. Схема пламенного фотометра. В качестве монохроматоров используют интерференционные светофильтры (Δλ ≈ 13 нм). Для поглощения постороннего излучения на пути светового потока ставят абсорбционные светофильтры. В атомно-эмиссионной спектроскопии используют прямоточные горелки с непосредственным введением аэрозоля (смесь раствора с воздухом) в пламени, а также горелки с предварительным смешиванием газов и аэрозоля (рис.2.15).  Рис. 2.15. Типы горелок для пламенной атомной спектроскопии: а - прямоточный горелка; б - горелка с предварительным смесителем топлива и пробы. Существуют одно - и много канальные устройства. Для одновременного определения двух или более элементов в одной пробе используют многоканальные устройства . 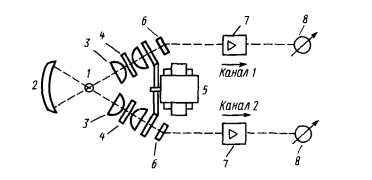 Рис. 2.16. Оптическая схема двухканального пламенного фотометра Flapho-4: 1 - горелка; 2 - зеркало; 3 - конденсорные линзы; 4 - интерференционные светофильтры; 5 - модулятор; 6 - селеновые фотоэлементы; 7 - усилители; 8 - гальванометры. Способы определения концентрации В эмиссионной фотометрии пламени для определения концентрации используют прямолинейную зависимость интенсивности аналитического сигнала (излучения) от концентрации раствора. Метод требует эталонов, то есть растворов с точно известной концентрацией. Обычно применяют метод градуировочного графика, строят в координатах сила фототока - концентрация. Вместо силы тока по оси ординат иногда откладывают условные единицы, определяются калибровкой шкалы устройства. Для этого устанавливают стрелку гальванометра на нуль при отсутствии элемента, который определяем в растворе и на 100% при максимальной концентрации эталонного раствора. Градуированный график может быть искаженным при очень низких концентрациях (имеет место самопоглощение света не возбужденными атомами). Если известен интервал линейной зависимости I от С, можно воспользоваться методом ограничивающих растворов. Для этого выбирают два эталоны (один из несколько меньшей c1, другой с большей c2 концентрацией, чем в исследуемом растворе cx) и измеряют их интенсивность I. cx=c1+ Если состав исследуемых образцов неизвестный отличается от стандартов (например, анализ природных объектов), то рекомендуется использовать метод добавок. При этом готовят три растворы (рис.2.17): первый - исследуемый раствор с концентрацией cx; второй - исследуемый раствор, в который добавлена известно количество стандартного раствора элемента, который определяется (cx + c1) третий - исследуемый раствор с добавкой стандартного раствора, примерно в 2 раза большей чем во второй (cx + c2). Строят график зависимости I (с поправкой на фон) от c, неизвестную концентрацию находят отрезком, который отсекается на отрицательной шкале абсцисс. 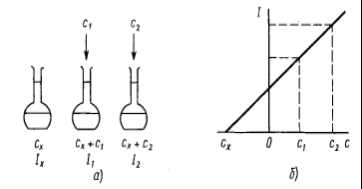 Рис. 2.17. Метод добавок: а - схема эксперимента; б - графическое определение концентрации. 2.3 Ионометрия. Особенности выбора и эксплуатации электродов Ионометрия имеет весьма ограниченную сферу применения, в основном в лабораторной практике. Объясняется это в первую очередь тем, что большинство ионоселективных электродов не обладают высокой избирательностью (селективностью). С другой стороны наиболее привлекательный метод - прямая потенциометрия не позволяет проводить анализ с высокой точностью, другие же потенциометрические методы более сложные и трудоемкие, а главное они не получили широкой популярности среди аналитиков. И третья особенность, ограничивает применение метода это довольно узкий диапазон определения, для большинства ионоселективных электродов составляет 4-6 порядков концентрации. Основная доля исследований в области ионоселективных электродов была проведена в 40-х ... 60-х годах 20-го века. С тех пор были отобраны лучшие электрохимические системы, на базе которых строят электроды. Все это объясняет тем, что подавляющее большинство ионоселективных электродов выпускаются в единственной модификации. Поэтому трудностей с выбором электродов обычно не возникает. Проблемы обычно возникают у новичков из-за недостаточного понимания специфики потенциометрического метода анализа. В первую очередь, перед тем как подбирать электроды, следует убедиться в применимости этого метода к конкретным условиям. В случае затруднений рекомендуется обратиться за консультацией к специалистам-аналитикам и разработчикам методик. 1 2 |