|
Смсм, однако этот факт устраняется путем введения определенных растворителей и солей в межмолекулярное пространство полимера
Оглавление
Введение
На сегодняшний день литий-ионные аккумуляторы считаются наиболее распространёнными перезаряжаемыми источниками энергии. Это обуславливается тем, что литий-ионным аккумуляторам свойственна высокая ионная и низкая электронная проводимость, а также широкое окно электрохимической стабильности. Типичный представитель данного класса химических источников тока имеет ионную проводимость порядка 10-2 См/см при комнатной температуре. Такая высокая ионная проводимость достигается за счет использования жидких электролитов, которые представляют собой раствор соли лития в различных смесях полярных апротонных органических растворителей. Однако, у жидких электролитов есть существенные недостатки, обусловленные низкой термостабильностью, воспламеняемостью, а также возможностью дендридообразованию в процессе работы. Эти факты наталкивают на мысль об использовании твердых неорганических электролитов, которые термоустойчивы и имеют высокую ионную проводимость порядка
10-2 См/см. Твердые неорганические электролиты лишены недостатков жидких, однако они имеют плохой контакт на межфазной границе электрод/электролит, что приводит значительному росту сопротивления. Мало того известно, что при заряде/разряде электроды меняют свой объем из-за интеркаляции лития. Твердотельные электролиты неспособны адаптироваться под такие изменения и по этой причине их не используют в литий-ионных аккумуляторах.
Достоинства жидких и твердых электролитов могут сочетать в себе полимерные электролиты. В таких электролитах полимер может выполнять роль матрицы, а его полярные группы переносят ионы по эстафетному механизму, координируясь вокруг них. Проводимость полимерных электролитов на несколько порядков ниже, чем у твердых неорганических (
10-6 См/см), однако этот факт устраняется путем введения определенных растворителей и солей в межмолекулярное пространство полимера.
Полимерные электролиты довольно эластичны: это решает проблему контакта электролита с электродом и расширяет диапазон конструктивных решений.
Обзор литературы 1.Полимерные электролиты
Полимерные электролиты характеризуются гибкостью, обеспечивающей удобную конструкцию аккумулятора. Наиболее распространенным классом твердых полимерных электролитов для ЛИА являются материалы, построенные на основе полимерных матриц с растворенными в них солями лития с объемным анионом (ClO4 – , PF6 – , BF4 – и др.) [12]. Для достижения максимальных показателей электропроводности в растворитель добавляют литиевые соли с концентрацией 1-1.5 моль на литр(М). Непременным условием для обеспечения растворимости этих солей является наличие в полимерной матрице электроотрицательных атомов, способных координировать ионы лития (рис. 1). Такими полимерами, например, являются полиэтиленоксид, различные поликарбонаты, полисилоксаны и др. [13]. Ионный перенос в основном реализуется в аморфных участках полимера подобных электролитов и обусловлен локальной сегментальной подвижностью [14].
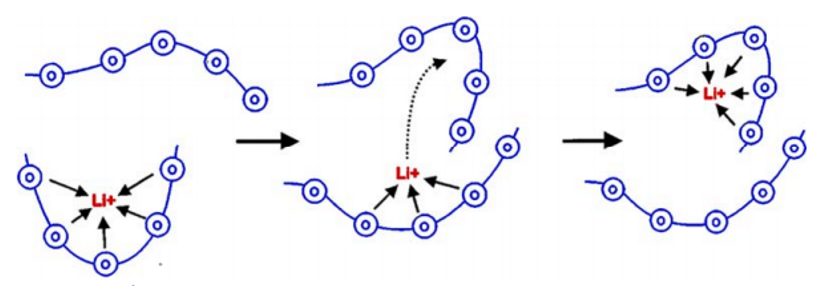
Рисунок 1. Транспортная модель переноса лития полимером.
Основными требованиями для электролитов служат: высокий диапазон рабочих температур, высокая ионная проводимость, высокая электрическая проницаемость(не менее 15 ед.), способность формирования защитных слоев у поверхности электродов, а также пассивирующих пленок на токоотводах во избежании коррозии. Согласно многочисленным экспериментам идеальных растворителей не существует. У одних чересчур высокая температура плавления, у других низкая темература кипения и т.д. Поэтому на практике принято либо их комбинировать между собой, либо брать за основу определенный вид растворителя и подбирать к нему соль для достижения ожидаемых результатов. Так, например пропиленкарбонат имеет температуру плавления -49 °С, и температуру кипения 242 °С. Такие показатели делают его привлекательным, например, по сравнению с растворителем диметилкарбонат, который имеет довольно высокую температуру плавления (5 °С) и низкую температуру кипения (91 °С). Однако, по сравнению с тем же диметилкарбонатом, пропиленкарбонат обладает высоким коэффициентом вязкости, что в свою очередь снижает транспорт ионов в объеме электролита. Поэтому, принято вводить дополнительные добавки в виде солей для увеличения электропроводности. Наиболее подходящей солью для данного растворителя является тетрафтороборат лития. Ионная группа BF4- имеет высокую ионную подвижность. В качестве электролита в ЛИА соль LiBF4 имеет определенные преимущества по сравнению с более распространенной солью LiPF6: она обладает большей термической стабильностью и влагоустойчивостью[ссылка]. Эксперименты показывают, что соль LiBF4 довольно устойчива к влаге при комнатной температуре, когда соль LiPF6 легко поддается гидролизву с образованием токсичных продуктов POF3 и HF при попадании эквивалентного количества воды. Такие побочные продукты приводят к разрушению электродных материалов батареи. Поэтому, с точки зрения безопасности сочетание пропиленкарбоната и тетрафторбората лития можно считать успешным.
В качестве перспективных электролитов для ЛИА можно рассматривать полимерные мембраны, содержащие ионогенные группы (SO3 – , COO– , [SO2 NSO2 CF3 ]– и др.) в литиевой форме [15, 16]. На сегодняшний день исследовано множество полимеров, однако мембрана Нафион, которую принято использовать в топливных элементах, обладает относительно высокой ионной проводимостью и её литиевую форму можно отнести к ион-селективным мембранам, поскольку она проводит исключительно ионы лития. Высокая ионная проводимость достигается лишь в сольватированной форме. Для возможности использования Нафиона в ЛИА в мембрану вводят безводные органические растворители, такие как линейные и/или циклические органические карбонаты, простые эфиры, диметилсульфоксид, диметилформамид [18, 20]. Таким образом Нафион может быть и электролитом и сепаратором одновременно.
Для использования Нафиона в литий-ионных аккумуляторах его необходимо перевести из кислой формы в литиевую. Это достигается путем выдержки мембраны в водном растворе LiOH (8 масс. %) при 60° С в течение 2-х часов, затем в 4 масс. % водном-спиртовом (вода – изопропанол = 1:1 по объему) растворе LiOH в течение 2-х часов при 60° С. После чего мембрану многократно промывают дистиллированной водой и высушивают в течение двух дней над P2O5 ( рис.2).
Для получения тонкопленочного электролита с пропиленкарбонатом в качестве пластификатора образцы литированной мембраны в условиях инертной атмосферы выдерживают в безводном пропиленкарбонате в течение двух дней при комнатной температуре.
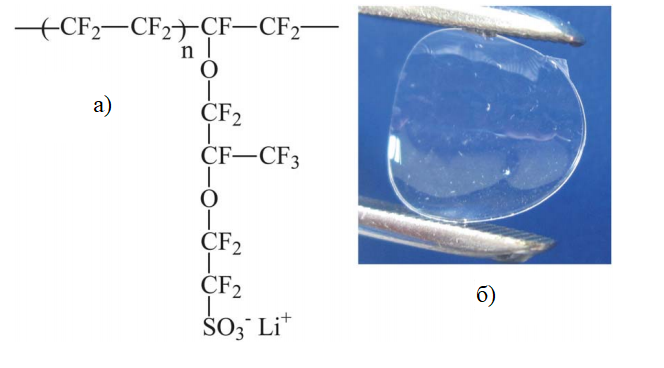
Рисунок 2. а) химическая структура литированной перфторированной мембраны Нафион; б) изображение литированной мембраны Нафион, которая впитала в себя растворитель PC (пропиленкарбонат).
Как отмечалось ранее, к достоинствам мембран типа Нафион относятся высокая ионная проводимость (достигающая при увлажнении рекордных значений до 10-1 См/см), прочность, химическая и термическая стабильность. По данным термического анализа, мембраны устойчивы 250–300 °С; при более высоких температурах начинаются процессы десульфирования и деструкции эфирных боковых цепей. Разложение основной полимерной цепи проходит при температурах выше 450 °С [21]. В будущем предполагается использование мембраны Нафион в качестве электролита и сепаратора в литий-серных аккумуляторах[ссылка]. Известно, что литий-серные аккумуляторы имеют более высокую ёмкость, превышающую ёмкость традиционных литий-ионных аккумуляторов в 2 раза. Однако литий-серные аккумуляторы имеют ряд недостатков, связанных с тем, что промежуточные продукты реакции – полисульфиды постепенно растворяются в электролите и мигрируют между катодом и анодом, способствуя высокому саморязряду. В таких аккумуляторах ион-селективная мембрана Нафион потенциально могла бы предотвратить излишние миграции ионных групп, повысив тем самым эффективность аккумулятора.
К недостаткам мембраны Нафион можно отнести её стоимость и малоизученность в области литиевых аккумуляторов.
2.Компьютерное моделирование
С развитием компьютерных технологий, вычислительная химия и физика стали мощными и необходимыми инструментами для исследования систем, в которых экспериментальные методы либо неэффективны, либо обеспечивают недостаточным объемом информации в связи с собственными ограничениями. Например, методы спектроскопии, несмотря на свою скорость, могут быть использованы для наблюдения быстрых химических реакций только на молекулярном уровне. Множество дифракционных методов применяются для получения подробной информации о кристаллической структуре, однако такие методы сопровождаются сложностью мониторинга изменений на молекулярном уровне. Поэтому, экспоненциальный рост вычислительной мощности приводит к соответствующему росту вычислительных методов в точных науках, таким как метод “из первых принципов”, полуэмпирические методы, теория функционала плотности, метод Монте-Карло, молекулярная механика, молекулярная динамика и т.д.
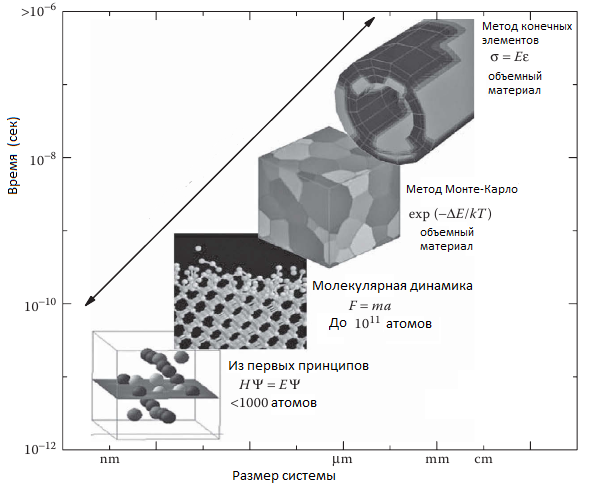
Рисунок 3. Компьютерные методы в вычислительной физике. По оси абсцисс отложен размер моделируемой системы, по оси ординат – время вычислительного эксперимента.
Компьютерное моделирование включает в себя следующие процедуры:
Определение типов физических величин или явлений, которые следует рассчитать.
Создание модели системы, которая корректно представляет реальную, существующую систему.
Выбор соответствующих правил (законов) для расчета системы (классическая механика, квантовая механика, теории и алгоритмы и т.д.)
Выбор кода/программы для выполнения моделирования.
Запуск моделирования и сравнение полученных данных с экспериментальными или рассчитанными в других работах.
Последний пункт является особенно важным, так как данный этап позволяет оценить правильность соблюдения всех предыдущих пунктов.
2.1 Метод молекулярной динамики
Метод молекулярной динамики может быть рассмотрен как очень полезный инструмент для изучения полимерных электролитов. Молекулы полимеров довольно велики для более сложных методов симуляции, использующих меньшее число приближений (напр. квантово-механические методы). Метод молекулярной динамики позволяет взглянуть на структурные и динамические свойства системы, а также оценить их взаимосвязь, информация о которой необходима для анализа ионного транспорта в системе. Более того, транспортные механизмы обычно не вовлекают в себя редокс-реакции, что безусловно позволяет проводить исследование ионного транспорта в рамках данного метода.
Суть данного метода довольно проста: молекулярная динамика рассматривает атомы как самостоятельные частицы и игнорирует электронные и ядерные взаимодействия (рис. 4) [1]. Таким образом, система, состоящая из таких частиц, может быть описана уравнениями движения Ньютона. Еще в 1960-х годах методом молекулярной динамики можно было смоделировать поведение всего лишь нескольких сотен атомов, когда в настоящее время эта цифра достигает 1011.
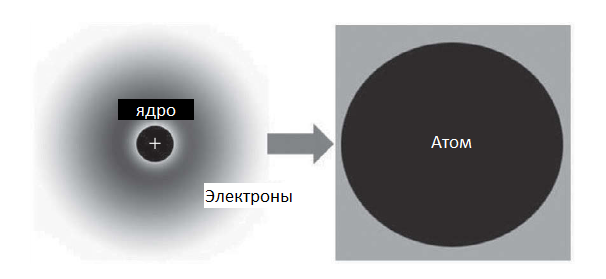
Рисунок 4. Схематичное представление атома в методе молекулярной динамики
Существуют следующие основные положения метода молекулярной динамики:
Для того чтобы описать частицы используется классическая механика Ньютона.
Длина волны Де Бройля атома (частицы) должна быть много меньше, чем расстояние между атомами.
Межатомные силы представляются в виде классических потенциальных сил (градиент потенциальной энергии системы).
Для того чтобы получить результаты макроскопического характера необязательно точное знание траектории движения частиц системы на больших интервалах времени.
Все конфигурации, получаемые в ходе вычислений методом молекулярной динамики, подчиняются статистической функции распределения.
В момент, когда атомы находятся достаточно близко друг от друга между ними возникает силы притяжения и отталкивания. Такое взаимодействие описывается межатомным потенциалом[1]. В конечном счете атомы примут равновесное состояние в минимуме потенциальной энергии. Сумма всех действующих сил равна:
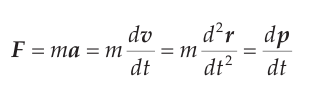 (1) (1)
где:
а – ускорение; v – скорость; t – время; r – радиус вектор; p – импульс
В случае изолированной системы силы F, действующих на атом можно представить как отрицательный градиент от потенциальной энергии взаимодействия атомов:
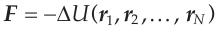 (2) (2)
В общем виде задачу молекулярной динамики можно описать следующим образом:
по известным радиус-векторам 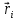 всех атомов системы в момент времени t в первую очередь вычисляется потенциальная энергия каждого атома Ui и находятся силы всех атомов системы в момент времени t в первую очередь вычисляется потенциальная энергия каждого атома Ui и находятся силы 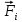 , которые на него действуют в данный момент времени. , которые на него действуют в данный момент времени.
зная известные силы, скорости и радиус-векторы находят новые скорости и радиус-векторы в момент времени 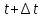 ; ;
новые значения радиус-векторов и скоростей сохраняются для дальнейшего анализа;
вновь происходит увеличение времени на 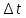 ; ;
если счетчик времени не достиг заданного времени окончания моделирования, то вычислительный эксперимент возвращается к пункту 1 для выполнения следующих шагов моделирования.
2.2 Потенциальная энергия взаимодействия атомов
Для примера, рассмотрим парные взаимодействия двух атомов. Число атомов в системе обозначим за N, тогда число (N – 1) это количество парных взаимодействий на один атом (рис 5.) [1]. Таким образом мы можем посчитать число пар по следующей формуле:
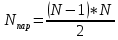 (3) (3)
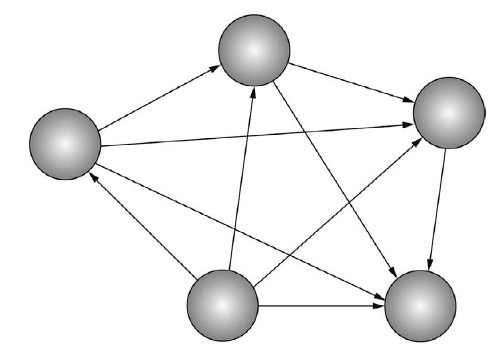
Рисунок 5. Парные взаимодействия (указаны стрелками) в системе, состоящей из пяти атомов.
Параметры такой простой системы, учитывающей только лишь парные взаимодействия, можно посчитать используя потенциал Леннарда – Джонса[4]. Потенциальная энергия взаимодействия атомов 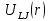 определяется следующим образом: определяется следующим образом:
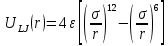 (4) (4)
где:
параметр 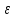 это минимальная энергия потенциальной кривой (когезионная энергия); это минимальная энергия потенциальной кривой (когезионная энергия);
параметр 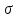 это межатомное расстояние, при котором потенциальная энергия равна нулю (рис. 6) [1]; это межатомное расстояние, при котором потенциальная энергия равна нулю (рис. 6) [1];
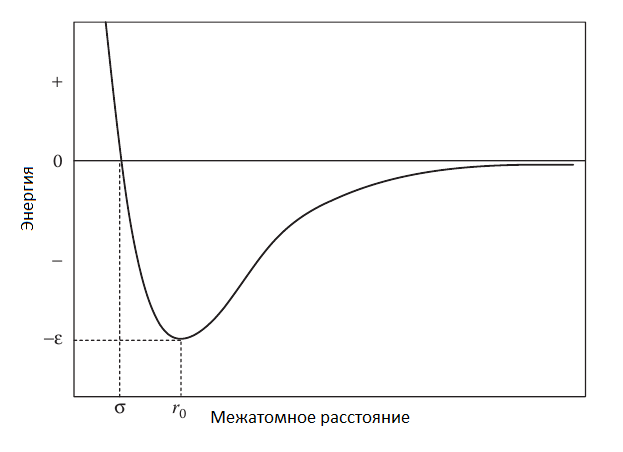
Рисунок 6. График зависимости потенциальной энергии от межатомного расстояния.
Важно отметить, что:
1.При расстояниях r 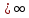 , потенциал равен нулю. , потенциал равен нулю.
2.Когда атомы сближаются, включается механизм диполь-дипольного взаимодействия и величина r-6 описывает Ван-дер-Ваальсовое взаимодействие(притяжение).
3.В момент, когда атомы слишком близки друг другу, включается механизм запрета Паули и величина r-12описывает отталкивание.
Потенциал Леннарда – Джонса[4] можно широко использовать для моделирования динамики инертных газов, однако многие атомные эффекты игнорируются и данный потенциал не может быть применим для металлов, полупроводников и прочих твердых материалов.
2.3 Алгоритм Верле
Существует множество способов и методов решения системы уравнений движения. Все эти методы стремятся улучшить расчеты и достичь высокой скорости работы. В молекулярной динамике чаще всего используют так называемый алгоритм Верле. Он имеет хорошие соотношения между точностью и скоростью. Метод Верле заключается в том, что позиции атомов (их координаты) сохраняются на целом шаге моделирования[1]:
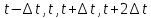 (5) (5)
и т.д, а скорости хранятся на половинном шаге
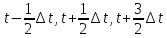 (6) (6)
и т.д.
Для того, чтобы рассчитать действующие на атом силы находятся производные потенциальной энергии:
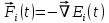 (7) (7)
После производится расчет новых скоростей на половинном шаге:
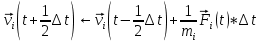 (8) (8)
Затем находятся скорости на целом шаге:
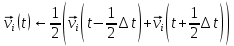 (9) (9)
Это делается для того, чтобы имелась возможность вычислить термодинамические свойства в момент времени t.
Новые координаты атомов определяются с помощью значений скорости на половинном шаге:
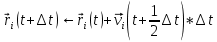 (10) (10)
2.4 Периодические граничные условия
Используя периодические граничные условия на межатомные взаимодействия, можно получить “бесконечное” множество частиц – объемную фазу вещества[1].
В процессе моделирования все молекулы или атомы размещаются в одной ячейке, имеющей форму параллелепипеда. Эта ячейка является центральной. Для того, чтобы создать условие, в котором исключается возможность появления атома или молекулы на границе данной ячейки, можно размножить эту ячейку с помощью трансляции по всем сторонам.
Для того, чтобы частицы не взаимодействовали сразу со всеми другими образами частиц одновременно, вводится радиус отсечения взаимодействий Rcutoff (рис. 7) [1]. По условию, такой радиус не должен превышать половину длины самого короткого ребра ячейки[1]:
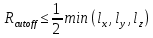 (11) (11)
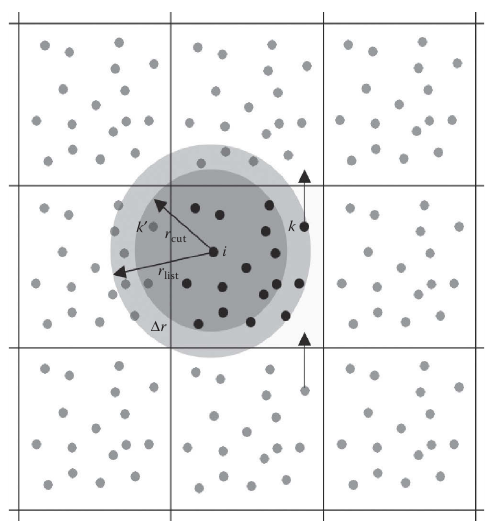
Рисунок 7. Периодические граничные условия для некоторой системы с применением радиуса отсечения взаимодействий. На рисунке rcut – радиус отсечения, rlist – радиус захвата ближайших соседей. k и k’ – атомы, попадающие в область, которая отсекается соответствующими радиусами.
Цели и задачи
Поскольку в полимерных электролитах всегда встаёт вопрос проводимости, то в данной работе было принято решение исследовать проводимость лития в мембране Нафион, используя метод молекулярной динамики в качестве инструмента для исследования. В соответствии с этим были сформулированы следующие задачи:
1.Построить расчетную систему и провести вычислительный эксперимент.
2.Оценить структурные особенности системы.
2.Оценить коэффициент диффузии лития.
3.Посчитать ионную проводимость лития.
4.Оценить плотность системы.
Методика исследования 1.Математическая модель.
За основу, описывающую потенциальную энергию системы, была принята следующая модель:
U=Ub+Uv+Uφ+Uqq+Uvdw (11)
где Ub – потенциальная энергия валентных связей, Uv – валентных углов, Uφ - торсионных углов, Uqq – кулоновских сил, Uvdw – взаимодействий Ван-дер-Ваальса.
Энергия валентных связей описывается параболическим потенциалом:
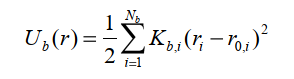 (12) (12)
где Kb,i – эффективная жесткость валентной связи, i – номер связи в молекуле, Nb – полное число валентных связей, ri – длина связи, r0,i – равновесная длина связи.
Потенциальная энергия валентных углов также представляется гармоническим потенциалом:
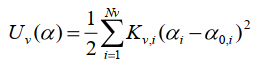 (13) (13)
где Kv,i – эффективная упругость валентного угла, i – номер валентного угла, Nv – полное число валентных углов, αi – значение валентного угла, α0,i – его равновесное значение.
Потенциальная энергия для торсионных углов представлена в виде:
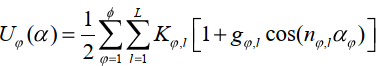 (14) (14)
где Kφ,l – константа, φ – номер торсионного угла, l – номер гармоники, gφ,l – вклад гармоники в потенциал торсионного угла (–1 < gφ,l < 1), nφ,l – кратность гармоники. Потенциалы Uf и Uφ отличаются константами. Потенциальная энергия взаимодействия заряженных атомов характеризуется кулоновским потенциалом:
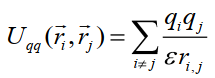 (15) (15)
где ri, rj – радиус-векторы взаимодействующих атомов, qi, qj – их парциальные заряды, ε –диэлектрическая проницаемость среды (для вакуума ε = 1), ri,j = | ri - rj | - расстояние между атомами.
Ван-дер-ваальсовские взаимодействия описываются потенциалом Леннарда-Джонса:
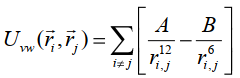 (16) (16)
где A и B– константы, определяющие глубину потенциальной ямы и расположение её минимума, ri, rj – радиус-векторы взаимодействующих атомов, ri,j = | ri - rj | - расстояние между атомами.
Параметры и коэффициенты силовых полей для каждой молекулы были сгенерированы с помощью веб-сервиса ATB(Automated Topology Builder)[22].
2.Детали проведения вычислительного эксперимента.
Согласно целям исследования, была построена система, содержащая в себе молекулы Нафиона, атомы лития, молекулы тетрофторбората (далее LiBF4) и пропиленкарбоната (далее PC) в качестве растворителя(рис.9). Общее количество атомов и молекул представлено в таблице 1.
Таблица 1. Данные о составе системы
Молекулы Нафиона, шт.
|
Атомы лития, шт.
|
Молекулы тетрофторбората, шт.
|
Молекулы пропиленкарбоната, шт.
|
Общее количество атомов, шт.
|
5
|
355
|
305
|
480
|
11520
|
Модель мономера Нафиона-117 представлена на рисунке 8. Согласно литературным источникам [21], эта модель является наиболее распространенной. Эквивалентный вес такой модели равен 1100 г/моль (n = 7). Для двух систем было сгенерировано 5 цепочек Нафиона, содержащие в себе по 10 мономеров (рис. 10)

Рисунок 8. Модель мономера Нафиона.
Для системы использовался растворитель PC в соотношении 1:1 по массе относительно молекул Нафиона, атомов лития и соли. Визуализация модели растворителя и соли представлена на рисунке 5.
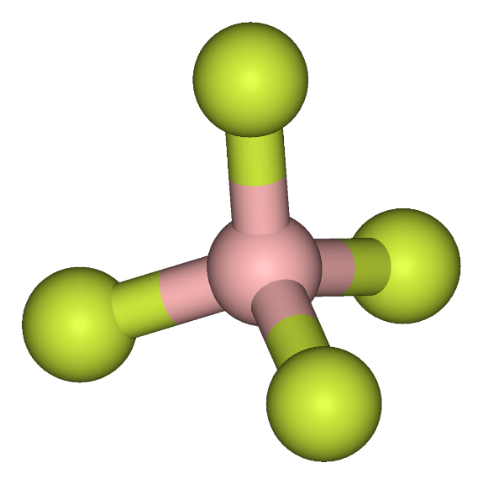
|
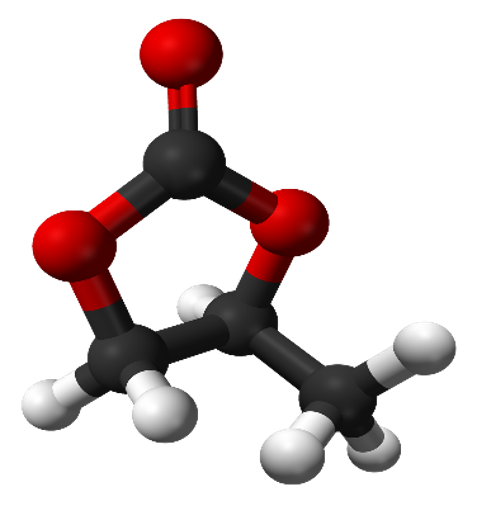
|
А
|
Б
|
Рисунок 9. А – тетрафторборат (BF4), Б – пропиленкарбонат (PC).
Взаимное расположение всех молекул для двух систем было сгенерировано случайным образом в виртуальном пространстве размером 300 х 300 х 300 Å (рис. 10). В процессе расчетов учитывались периодические граничные условия. Радиус обрезки равен 14 Å. Временной шаг интегрирования равен 1 фс. Вычислительный эксперимент проводился в условиях изотермально-изобарического (т.е число частиц, давление и температура постоянны) и канонического ансамблей(т.е число частиц, объем и температура постоянны) с использованием термостата Нойза-Хувера. Внешнее давление равнялось 1 атм.
Во время формирования вычислительной коробки редко случается так, чтобы атомы и молекулы оказывались сразу в положении равновесия. Однако, содержимое коробки обязано прийти к состоянию равновесия перед тем, как начнется фиксация статистических состояний. Это особенно важно для таких сложных систем, как полимеры, которые имеют тенденцию “запутываться” во время случайной генерации их положения. Равновесие контролируется энергией системы, которая должна быть стабильна на момент начала отбора данных. Для достижения правдоподобных показателей равновесия, эксперимент для каждой системы выполнялся в четыре этапа: 1 - установление температуры 300 К в условиях изотермально-изобарического ансамбля с последующей выдержкой в течении 2 нс; 2 - нагрев системы до 400 К и выдержка в течении 2 нс; 3 – последующее охлаждение до 300 К в течении 2 нс. 4 – смена типа ансамбля на канонический с последующий выдержкой в течении 50 нс при температуре в 300 К. Согласно литературным источникам [23], такой вид термообработки позволяет достичь величины плотности, приближенной к реальной плотности растворителя.
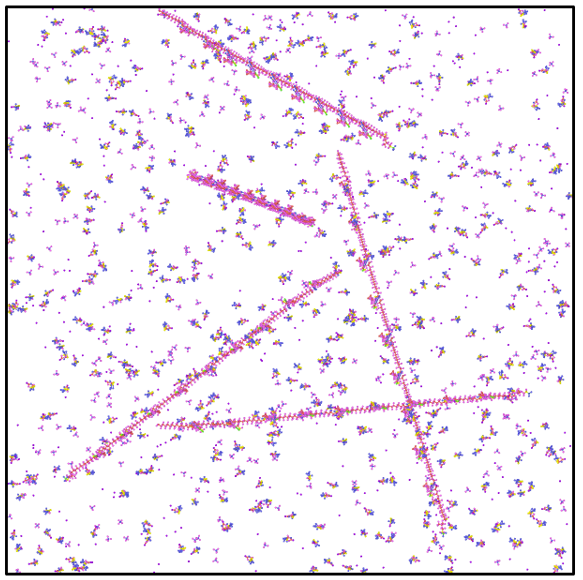
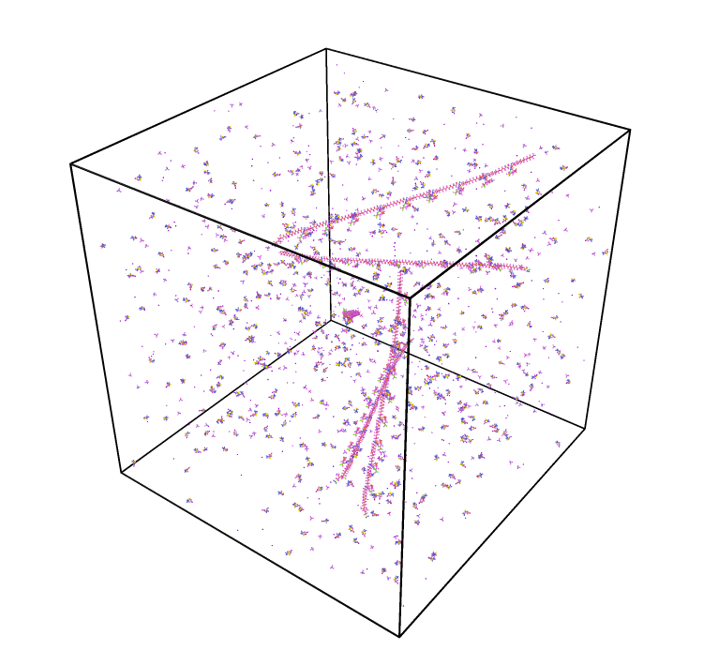
Рисунок 10. Первоначальный неоптимизированный вид системы. А – вид сверху, Б – трехмерная проекция.
Для проведения молекулярной динамики использовался программный пакет LAMMPS, разработанный австрийскими разработчиками [24]. Данное ПО устанавливалось на вычислительный ресурс, предоставляемый компанией Google под названием Google Colaboratory [25]. Облачные вычисления данного сервиса осуществляются на дистрибутиве Linux с использованием оболочки Jupyter Notebook, которая в свою очередь предоставляет возможность комбинирования языка программирования Python c командами Linux. Для моделирования молекул и их визуализации использовались программы VMD[26] и Avogadro[27]. Анализ полученных результатов осуществлялся с использованием языка программирования Python 3-й версии и библиотеки MDAnalysis[28].
3.Статистический анализ
После проведения вычислительного эксперимента используется статистический анализ для расчета различных свойств, связанные со структурой и динамическим поведением. Статистический анализ и его интерпретация составляют основу для дальнейших выводов о проделанной работе. Одним из самых важных свойств, которое можно получить из результатов молекулярной динамики является функция радиального распределения. Эта функция представляет собой вероятность нахождения частицы типа B на расстоянии r от частица типа A. В кристаллах эта функция представляет из себя кривую с периодически повторяющимися пиками. Такое распределение дает информацию как и о ближнем порядке, так и о дальнем одновременно. В жидкостях или в полимерных системах присутствует только ближний порядок: последующие небольшие пики на небольшом расстоянии соответствуют первой координационной сфере.
Функция радиального распределения задается следующим образом:
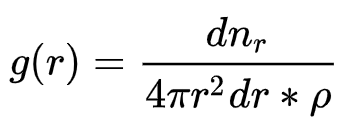 (17) (17)
где:
nr – количество частиц, чьи центры попали в область dr относительно атома красного цвета (см. рис.11). Величина dr подбирается экспериментально и как правило, чем она меньше, тем точнее расчет. В нашем случае, dr составляет 0,1 Å.
r – радиус сферы (радиус обрезки);
p – плотность системы.
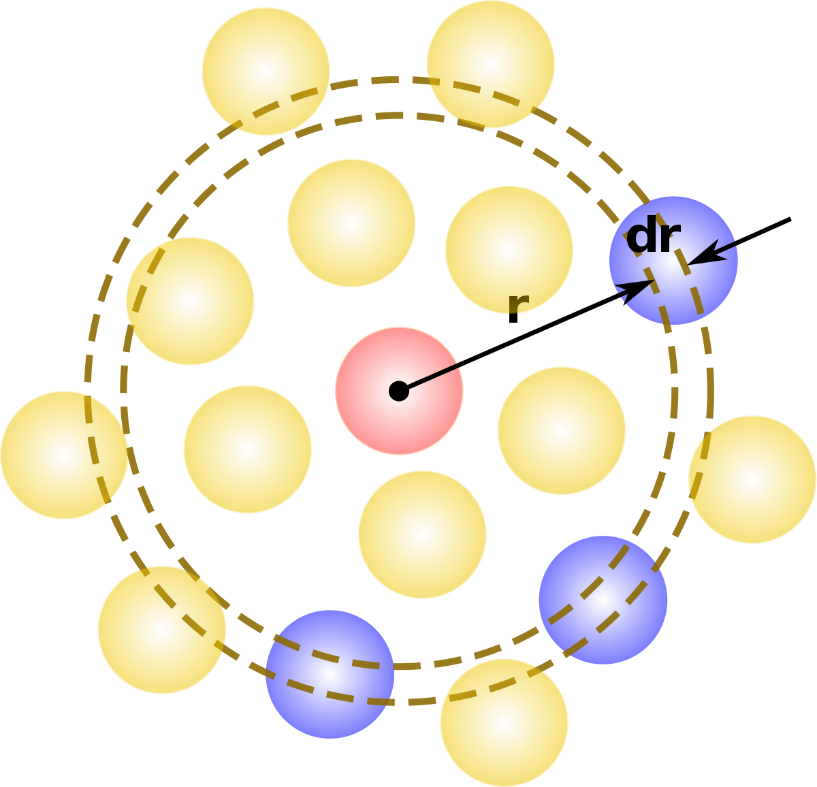
Рисунок 11. Количество частиц(отмечены синим цветом), попавших в область dr относительно атома, который отмечен красным цветом.
Помимо этого, для определения коэффициента диффузии необходимо вычислить среднеквадратичное смещение частиц относительно эталонного положения в момент времени t0 и получить кривую во времени.Величина среднеквадратичного смещения определяется следующим выражением:
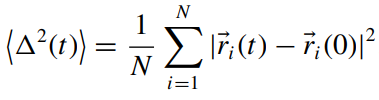 (18) (18)
где:
t - время;
N - общее число частиц;
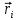 - радиус вектор i-ой частицы. - радиус вектор i-ой частицы.
Известно, что коэффициент диффузии пропорционален наклону прямой, полученной методом линейной регрессии кривой среднеквадратичного смещения. Данный факт описывается уравнением:
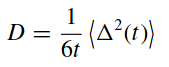 (19). (19).
Результаты и обсуждение 1.Функции радиального распределения
Графики для разных атомных групп представлены на рисунках 12 и 14.
Из графика А на рис.12, который определяет функцию радиального распределения для атомной группы “литий – SO3-“, можно заметить, что амплитуда самого узкого пика расположена на расстоянии 1,75 Å и она соответствует первой координационной сфере. Это в свою очередь демонстрирует тот факт, что ионы лития сильно связны с ионами SO3- группы и вероятность нахождения лития максимальна в этом случае. Затухающие, более широкие осцилляции соответствуют свободным ионам лития. Координационное число атомов лития в области молекулы SO3- составляет 4.
Помимо SO3- групп, также высока вероятность встретить литий в области молекулы BF4, что подтверждается на графике Б из рисунка 12. Пик этого графика расположен на расстоянии 1,96 Å. Координационное число атомов лития в области молекулы BF4 составляет 7.
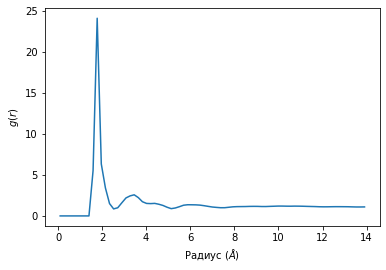 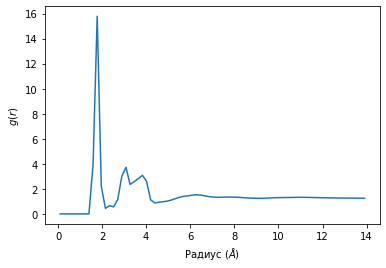
А Б
Рисунок 12. Функции радиального распределения. А- распределение иона лития относительно SO3- группы; Б – распределение иона лития относительно молекулы BF4.
Также, дополнительно были подсчитаны функции радиального распределения в областях PC молекулы, которые представлены на рисунке 13.
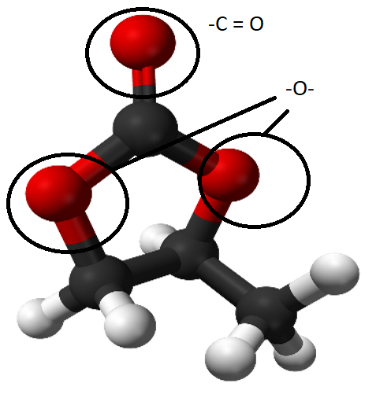
Рисунок 13. Области молекулы PC, вблизи которых рассчитывались функции радиального распределения по литию.
Графики радиального распределения лития по областям С=O и -О- представлены на рисунке 14. Анализ показал, что координационное число в области С=O равно 2, а в области -О- меньше единицы. Это свидетельствует о том, что литий слабо сольватирован молекулой пропиленкарбоната.
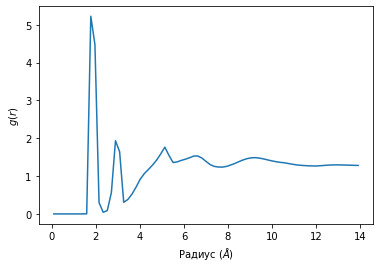 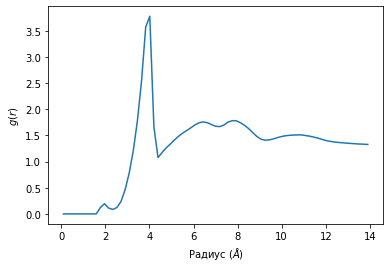
А Б
Рисунок 14. Функции радиального распределения. А - распределение иона лития относительно С=O группы; Б – распределение иона лития относительно -О- области.
Координационное число атомов лития относительно атомных групп рассчитывалось путем интегрирования функции радиального распределения в области первого пика:
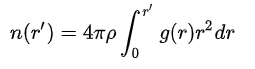 (20) (20)
где:
n(r’) - координационное число;
Результаты расчетов представлены в таблице 2:
Таблица 2. Координационные числа число атомов лития относительно атомных групп, полученные путем интегрирования функции радиального распределения в области первых пиков.
SO3-
|
BF4
|
-O-
|
-C=O
|
7
|
4
|
0.1
|
2
|
Анализ графиков А и Б из рисунков 12 и 14, а также подсчет координационных чисел в области первых пиков подтверждают факт того, литий чаще всего встречается у атомных групп SO3- и BF4. и слабо проявляется в области PC молекулы. Однако, проинтегрировав функцию радиального распределения в области от 7 до 14 Å становится возможна оценка доли свободных и связанных ионов лития. Интегрирование графиков и усреднение полученных значений показывает, что доля свободных ионов лития составляет 0,83 %. Этот факт учитывается в дальнейшем для оценки ионной проводимости.
2.Расчет коэффициента диффузии лития.
Для расчета коэффициентов диффузии были получены графики среднеквадратичного смещения для ионов лития и атомной группы SO3-. Графики представлены на рисунке 15.
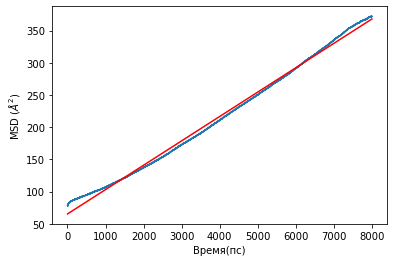
А
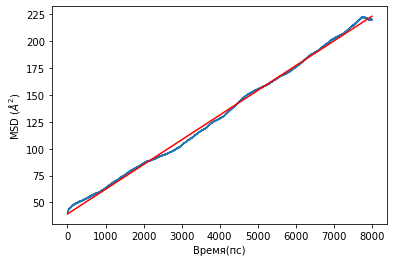
Б
Рисунок 15.Графики среднеквадратичного смещения для ионов лития для: А- для ионов лития, B- для группы SO3-.
Результаты коэффициентов диффузии представлены в таблице 3.
Растворитель
|
DLi
( * 10-7 cm2 c-1)
|
D SO3-
( * 10-7 cm2 c-1)
|
DfLi
( * 10-7cm2 c-1)
|
PC
|
6.3
|
3,8
|
6,8
|
*DMSO
|
6.0
|
3,9
|
7,2
|
*данные по этому растворителю приведены в работе[23]
|
Помимо коэффициентов, полученных в текущей работе были приведены данные из похожей работы [23], в которой оценивалась ионная проводимость лития в мембране Нафион с использованием диметилсульфоксида в качестве растворителя. Это было сделано для того, чтобы оценить как минимум порядок полученных расчетов, который в свою очередь успешно совпал. Согласно этому же литературному источнику[23], коэффициент диффузии для связанных атомов лития совпадает с коэффициентом диффузии атомной группы SO3-. Поэтому, коэффициент диффузии свободных атомов лития DfLi рассчитывался согласно следующему выражению:
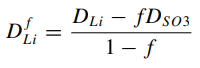 (21) (21)
где:
DcLi - коэффициент диффузии для связанных атомов лития(он же D SO3-);
f - доля связанных ионов лития.
3.Расчет ионной проводимости.
Информация приведенная в таблице 3 используется для расчета ионной проводимости лития, которая в свою очередь рассчитывается из уравнения Нернста-Эйнштейна:
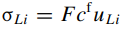 (22) (22)
где:
σLi - ионная проводимость;
F - постоянная Фарадея;
Cf – концентрация свободных ионов лития. Cf = С(1 - f);
ULi – подвижность иона лития.
Подвижность в свою очередь определяется следующим образом:
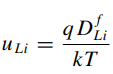 (23) (23)
где:
q - элементарный заряд;
k - постоянная Больцмана;
T - температура;
Полученные результаты и их сравнение с экспериментальными данными приводится в таблице 4.
Таблица 4.Расчеты ионной проводимости, подвижности и плотности в сравнении с экспериментальными данными их литературных источников.
σLi (* 10-4 См*см-1)
|
ULi (Кл * с*кг-1)
|
p(г*мЛ-1)
|
6.2
|
2.631 * 10-5
|
1.11
|
2[31]*
|
-
|
-
|
6.9[30]*
|
-
|
-
|
2,02[29]*
|
-
|
1,21[29]*
|
* Экспериментальные данные
|
Из таблицы 4 видно, что данные, полученные в настоящей работе хорошо коррелируют с экспериментальными данными из различных литературных источников. Согласно им, можно считать что ионная проводимость лития в текущей системе была оценена корректно и в последствии любой другой статистический анализ данной системы позволит изучить механизмы диффузии лития более детально с высокой степенью достоверности.
Вывод.
В ходе проведения вычислительных экспериментов было установлено следующее:
1.Из графиков радиального распределения выявлено, что ионы лития в основном координируются вокруг ионных групп SO3-(коорд.число = 7) и BF4--(коорд.число = 4).У молекул пропиленкарбоната ионы лития координируются слабее(2 атома в области -C=O связи).
2.Коэффициенты диффузии, полученные в данной работе хорошо соотносятся с данными из литературных источников. Так, для доли свободных ионов лития он составляет 6,8 * 10-7cm2 c-1, для связанных ионов 3,8 * 10-7cm2 c-1
3.Оценены ионная проводимость и плотность полученной системы. Ионная проводимость лития составляет 6,2 * 10-4 См*см-1, плотность системы 1,11 г*мЛ-1. Результаты хорошо коррелируют с экспериментальными данными из литературных источников. Согласно им, можно считать, что ионная проводимость лития в текущей системе была оценена корректно и в последствии любой другой статистический анализ данной системы позволит изучить механизмы диффузии лития более детально с высокой степенью достоверности.
|
|
|
 Скачать 2.19 Mb.
Скачать 2.19 Mb.