BKh_последняя 14. 1. Кровь. Понятие, физиологические функции
 Скачать 1.51 Mb. Скачать 1.51 Mb.
|

|
Тканевый активатор плазминогена (ТАЛ) - протеолитический фермент, содержащийся в эндотелии сосудов всех тканей, кроме печени. Поступление этого активатора в кровь увеличивается при эмоциональном напряжении, боли, венозной тромбоэмболии, умеренной физической работе. ТАЛ частичным протеолизом превращает неактивный плазминоген в активный плазмин. Активаторами плазминогена также служат фактор ХIIа и калликреин. Растворение фибринового сгустка происходит при взаимодействии фибрина, плазминогена и ТАП (рис. 14-18). Формирование сети фибриновых волокон при образовании тромба сопровождается сорбцией на ней плазминогена и его активаторов. В молекуле плазмина и плазминогена есть участки, комплементарные доменам фибрина, причём одна молекула плазмина может связывать несколько молекул фибрина. Молекулы ТАП тоже имеют центры связывания с фибрином. Образующийся из плазминогена под действием ТАП плазмин гидролизует фибрин с образованием пептидов X и Y, активирующих фибринолиз, и пептидов D и E, его тормозящих. Растворимые пептиды X, Y, D, E поступают в кровоток и там фагоцитируются. Разрушение тромба приводит к освобождению из него плазмина и ТАП. В кровяном русле последние быстро инактивируются специфическими ингибиторами и улавливаются печенью. ТАП ингибируется ингибиторами тканевого активатора плазмина первого (и-ТАП-1) и второго (и-ТАП-2) типов, а плазмин - α2-антиплазмином или другими ингибиторами сериновых протеаз. В почках синтезируется протеолитический активатор плазминогена урокиназа, которая, превращая плазминоген в плазмин, способствует освобождению почечных клубочков от фибриновых волокон. 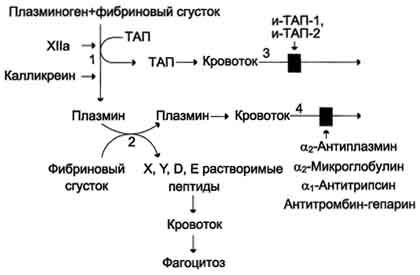 Рис. 14-18. Схема фибринолиза. 1 - абсорбированный на фибриновом сгустке плазминоген под действием активаторов (фактор ХIIа, калликреин, ТАП) частичным протеолизом превращается в плазмин; 2 - плазмин гидролизует фибрин с образованием растворимых пептидов X, Y,D, E; 3 - в кровотоке ТАП инактивируется специфическими белками и-ТАП-1, и-ТАП-2; 4 - активность плазмина снижается под действием неспецифических ингибиторов сериновых протеаз (α2-антиплазмина, α2-макроглобулина, α1-антитрипсина, комплекса антитромбин-гепарин). Из β-гемолитического стрептококка выделили белок стрептокиназу, образующий комплекс с плазминогеном, в котором плазминоген аутокаталитически превращается в плазмин. Урокиназу, стрептокиназу и ТАП используют при тромболитической терапии инфаркта миокарда, тромбозах вен и артерий, гемодиализе. Такие ингибиторы ферментов свёртывания крови, как α2-макроглобулин, α1-антитрипсин и комплекс антитромбин III-гепарин также обладают небольшой фибринолитической активностью. Снижение фибринолитической активности крови сопровождается тромбозами. Нарушение разрушения фибринового сгустка может быть вызвано наследственным дефицитом плазмино-гена или генетическим дефектом его структуры, снижением поступления в кровь активаторов плазминогена, повышением содержания в крови ингибиторов фибринолиза (и-ТАП-1, и-ТАП-2, α2-антиплазмина). Наследственные и приобретённые нарушения гемостаза могут привести как к геморрагическим заболеваниям, характеризующимся кровоточивостью, так и к тромботической болезни. Однако следует отметить, что повышенная склонность к тромбообразованию и внутрисосудистому свёртыванию (тромбофилии) встречается гораздо чаще, чем гемофилии. Например, частота разных форм гемофилии колеблется в разных странах от 6 до 18 на 100 000 мужчин, в то время как тромбофилии, вызванные дефицитом антитромбина III, встречаются у 1-2 больных на 5000, а при недостатке протеина С - у одного на 15 000 человек. Вопрос 28. Нарушения процессов свертывания крови. Тромботические и геморрагические состояния. ДВС – синдром. (инфа из интернета) КОАГУЛОПАТИИ Коагулопатии (coagulopathiae) — патологические состояния, связанные с нарушением в системе свертывания крови, приводящие обычно к развитию геморрагического синдрома. Коагулопатии делятся на: наследственные; приобретенные. Наследственные коагулопатии вызваны генетически обусловленным снижением количества плазменных компонентов гомеостаза или их некачественностью. Самые распространенные формы коагулопатии – афибриногенемия и гемофилии А, В, С. Наследственные коагулопатии делятся на: Гемофилии: А-дефицит VIII фактора, В-дефицит IX фактора, С-дефицит XI фактора, Д-дефицит XII; Парагемофилия: дефицит II, V, VII, X факторов; Нарушение образования фибрина, дефицит фибриногена (I фактора). Приобретенные коагулопатии вызываются инфекционными заболеваниями, тяжелыми энтеропатиями, болезнями печени и почек, геморрагическими васкулитами, злокачественными опухолями и другими заболеваниями. Коагулопатия может возникнуть в результате химических, механических, фармацевтических и физических воздействий. Одним из видов коагулопатий является гемодилюционная коагулопатия, она развивается у больных с острой кровопотерей, вызванной потерей белка или клеток крови, в первую очередь тромбоцитов. Клинические проявления коагулопатии: бледность кожи; геморрагический синдром; кровоизлияния в мягкие ткани, вызывающие обширные гематомы; гамартрозы; внутренние кровоизлияния; гематурия. Тромботические сосотояния- это патологическое состояние, характеризующееся нарушением системы свёртываемости крови при которой увеличивается риск развития тромбоза. ДВС-синдром – сидром дессиминированного внутрисосудистого свертывания. Фазы : I фаза — гиперкоагуляция. Потеря факторов свертывающей системы в процессе обильного кровотечения приводит к удлинению времени образования сгустка и его ретракции, удлинению времени капиллярного кровотечения. Лабораторные показатели: уменьшение времени свертывания крови, тромбинового времени, положительный этаноловый тест. II фаза — гипокоагуляция. При геморрагическом шоке в фазе спазма венул и артериол (клинические проявления: дегидратация, бледные и холодные кожные покровы, признаки острой почечной недостаточности) в капиллярах развивается расслоение плазмы и форменных элементов — “сладж”-феномен. Агрегация форменных элементов, обволакивание их фибрином сопровождаются потреблением факторов свертывания крови и активацией фибринолиза. Лабораторные показатели: умеренная тромбоцитопения (до 120 × 10^9/л), тромбиновое время 60 с и больше, резко положительный этаноловый тест.’ III фаза — потребления с активацией местного фибринолиза. Афибриногенемия в сочетании с выраженной активацией фибринолиза. При этой фазе рыхлые сгустки крови в месте кровотечения быстро (в течение 15-20 мин) лизируются на 50 %. Лабораторные показатели: увеличение времени свертывания крови, тромбинового времени, уменьшение тромбоцитов до 100 × 10^9/л, быстрый лизис сгустка. IV фаза — генерализованный фибринолиз. Капиллярная кровь не свертывается, отмечаются паренхиматозное кровотечение, петехиальные высыпания на коже и внутренних органах, гематурия, выпот в синовиальные полости и терминальные изменения в органах и системах. Причины: многие заболевания, сопровождающиеся разрушением эритроцитов, лейкоцитов, тромбоцитов и тканей или экспрессией тканевого фактора стимулированными эндотелиальными клетками, мооцитами и макрофагами, при переливании несовместимой крови, травмы, ожоги и обморожения, инфекционные и онкологические заболевания. Вопрос 29. Остаточный азот крови. Понятие, компоненты, содержание в норме. Азотемия, типы, причины возникновения. Остаточный азот (небелковый) - остающимся в фильтрате после осаждения белков. В состав небелкового азота входит главным образом азот конечных продуктов обмена простых и сложных белков. Небелковый азот крови включает азот мочевины (50% от общего количества небелкового азота), аминокислот (25%), эрготионеина (8%), мочевой кислоты (4%), креатина (5%), креати-нина (2,5%), аммиака и индикана (0,5%) и других небелковых веществ, содержащих азот (полипептиды, нуклеотиды, нуклеозиды, глутатион, билирубин, холин, гистамин и др.) У здорового человека колебания в содержании небелкового (остаточного) азота крови незначительны и в основном зависят от количества поступающих с пищей белков. При ряде патологических состояний уровень небелкового азота в крови повышается. Это состояние носит название азотемии. Азотемия в зависимости от вызывающих ее причин подразделяется на ретенционную и продукционную. Ретенционная азотемия развивается в результате недостаточного выделения с мочой азотсодержащих продуктов при нормальном поступлении их в кровяное русло. Она в свою очередь может быть почечной и внепочечной. При почечной ретенционной азотемии концентрация остаточного азота в крови увеличивается вследствие ослабления очистительной (экскреторной) функции почек. Резкое повышение содержания остаточного азота происходит в основном за счет мочевины. В этих случаях на долю азота мочевины приходится 90% небелкового азота крови вместо 50% в норме. Внепочечная ретенционная азотемия может возникнуть в результате тяжелой недостаточности кровообращения, снижения артериального давления и уменьшения почечного кровотока. Нередко внепочечная ретенционная азотемия является результатом наличия препятствия оттоку мочи после ее образования в почке. Продукционная азотемия развивается при избыточном поступлении азотсодержащих продуктов в кровь, как следствие усиленного распада тканевых белков при обширных воспалениях, ранениях, ожогах, кахексии и др. Нередко наблюдаются азотемии смешанного типа. В количественном отношении главным конечным продуктом обмена белков в организме является мочевина. Принято считать, что мочевина в 18 раз менее токсична, чем остальные азотистые вещества. При острой почечной недостаточности концентрация мочевины в крови достигает 50–83 ммоль/л (норма 3,3–6,6 ммоль/л). Нарастание содержания мочевины в крови до 16–20 ммоль/л (в расчете на азот мочевины) является признаком нарушения функции почек средней тяжести, до 35 ммоль/л – тяжелым и свыше 50 ммоль/л – очень тяжелым нарушением с неблагоприятным прогнозом. Иногда определяют отношение азота мочевины крови к остаточному азоту крови (в процентах): В норме это соотношение меньше 48%. При почечной недостаточности оно повышается и может достигать 90%, а при нарушении мочевинообразо-вательной функции печени снижается (ниже 45%). К важным небелковым азотистым веществам крови относится также мочевая кислота. Напомним, что у человека мочевая кислота является конечным продуктом обмена пуриновых оснований. В норме концентрация мочевой кислоты в цельной крови составляет 0,18–0,24 ммоль/л (в сыворотке крови – около 0,29 ммоль/л). Повышение содержания мочевой кислоты в крови (гиперурикемия) – главный симптом подагры. При подагре уровень мочевой кислоты в сыворотке крови возрастает до 0,5–0,9 ммоль/л и даже до 1,1 ммоль/л. В состав остаточного азота входит также азот аминокислот и полипептидов. В крови постоянно содержится некоторое количество свободных аминокислот. Часть из них экзогенного происхождения, т.е. попадает в кровь из пищеварительного тракта, другая часть аминокислот образуется в результате распада белков ткани. Почти пятую часть содержащихся в плазме аминокислот составляют глутаминовая кислота и глутамин . Содержание свободных аминокислот в сыворотке и плазме крови практически одинаково, но отличается от уровня их в эритроцитах. В норме отношение концентрации азота аминокислот в эритроцитах к со- держанию азота аминокислот в плазме колеблется от 1,52 до 1,82. Это отношение отличается большим постоянством, и только при некоторых заболеваниях наблюдается его отклонение от нормы. Вопрос 30. Обмен железа: всасывание, транспорт кровью, депонирование. Роль железа в процессах жизнедеятельности. Железо. Норма 3-4 г. В гемсодержащих белках железо находится в составе гема. В негемовых железосодержащих белках железо непосредственно связывается с белком. К таким белкам относят трансферрин, ферритин, окислительные ферменты рибонук-леотидредуктазу и ксантиноксидазу, железофлавопротеины NADH-дегидрогеназа и сукцинат-дегидрогеназа. В нейтральной или щелочной среде железо находится в окисленном состоянии - Fe3+, образуя крупные, легко агрегирующие комплексы с ОН-, другими анионами и водой. При низких значениях рН железо восстанавливается и легко диссоциирует. Процесс восстановления и окисления железа обеспечивает его перераспределение между макромолекулами в организме. Ионы железа обладают высоким сродством ко многим соединениям и образуют с ними хелатные комплексы, изменяя свойства и функции этих соединений, поэтому транспорт и депонирование железа в организме осуществляют особые белки. В клетках железо депонирует белок ферритин, в крови его транспортирует белок трансферрин. Всасывание железа в кишечнике В пище железо в основном находится в окисленном состоянии (Fe3+) и входит в состав белков или солей органических кислот. Освобождению железа из солей органических кислот способствует кислая среда желудочного сока. Наибольшее количество железа всасывается в двенадцатиперстной кишке. Аскорбиновая кислота, содержащаяся в пище, восстанавливает железо и улучшает его всасывание, так как в клетки слизистой оболочки кишечника поступает только Fe2+. В суточном количестве пищи обычно содержится 15 - 20 мг железа, а всасывается только около 10% этого количества. Организм взрослого человека теряет около 1 мг железа в сутки. Количество железа, которое всасывается в клетки слизистой оболочки кишечника, как правило, превышает потребности организма. Поступление железа из энтероцитов в кровь зависит от скорости синтеза в них белка апоферритина. Апоферритин "улавливает" железо в энтероцитах и превращается в ферритин, который остаётся в энтероцитах. Таким способом снижается поступление железа в капилляры крови из клеток кишечника. Когда потребность в железе невелика, скорость синтеза апоферритина повышается. Постоянное слущивание клеток слизистой оболочки в просвет кишечника освобождает организм от излишков железа. При недостатке железа в организме апоферритин в энтероцитах почти не синтезируется. Железо, поступающее из энтероцитов в кровь, транспортирует белок плазмы крови трансферрин. В плазме крови железо транспортирует белок трансферрин. Трансферрин - гликопротеин, который синтезируется в печени и связывает только окисленное железо (Fe3+). Поступающее в кровь железо окисляет фермент ферроксидаза, известный как медьсодержащий белок плазмы крови церулоплазмин. Одна молекула трансферрина может связать один или два иона Fe3+, но одновременно с анионом СО32- с образованием комплекса трансферрин-2 (Fe3+-CO32-). В норме трансферрин крови насыщен железом приблизительно на 33%. Трансферрин взаимодействует со специфическими мембранными рецепторами клеток. В результате этого взаимодействия в цитозоле клетки образуется комплекс Са2+-кальмодулин-ПКС, который фосфорилирует рецептор трансферрина и вызывает образование эндосомы. АТФ-зависимый протонный насос, находящийся в мембране эндосомы, создаёт кислую среду внутри эндосомы. В кислой среде эндосомы железо освобождается из трансферрина. После этого комплекс рецептор - апотрансферрин возвращается на поверхность плазматической мембраны клетки. При нейтральном значении рН внеклеточной жидкости апотрансферрин изменяет свою конформацию, отделяется от рецептора, выходит в плазму крови и становится способным вновь связывать ионы железа и включаться в новый цикл его транспорта в клетку. Железо в клетке используется для синтеза железосодержащих белков или депонируется в белке ферритине. Ферритин состоит из тяжёлых (21 кД) и лёгких (19 кД) полипептидных цепей, составляющих 24 протомера. Разный набор прогомеров в олигомере ферритина определяет образование нескольких изоформ этого белка в разных тканях. Ферритин представляет собой полую сферу, внутри которой может содержаться до 4500 ионов трёхвалентного железа, но обычно содержится менее 3000. Тяжёлые цепи ферритина окисляют Fe2+ в Fe3+, Железо в виде гидроксидфосфата находится в центре сферы, оболочка которой образована белковой частью молекулы. Оно поступает внутрь и освобождается наружу через каналы, пронизывающие белковую оболочку апоферритина, но железо может откладываться и в белковой части молекулы ферритина. Ферритин содержится почти во всех тканях, но в наибольшем количестве в печени, селезёнке и костном мозге. Незначительная часть ферритина экскретируется из тканей з плазму крови. Поскольку поступление ферэитина в кровь пропорционально его содержанию в тканях, то концентрация ферритина в крови - важный диагностический показатель запасов железа в организме при железодефидитной анемии. Роль: 1)Входит в состав гемоглобина(перенос кслорода) 2)Входит в состав миоглобина (создания кислородного запаса в организме. Благодаря наличию этих запасов можно, к примеру, нырнуть в воду и в течение какого-то времени не дышать, используя собственные запасы кислорода, накопленные благодаря железу миоглобина.) 3) Железо входит в структуру цитохромов, которые участвуют в процессах накопления энергии, выделяющейся во время заключительных этапов биологического окисления 4) Железо вместе с тем защищает органы от вредного воздействия токсичной перекиси водорода, продуцирующейся белыми кровяными клетками – лейкоцитами. В каталазе содержится железо, в присутствии которого молекулы перекиси водорода расщепляются на кислород и воду. 31. Тетрагидрофолиевая кислота, роль в синтезе и использовании одноуглеродных радикалов. Метилирование гомоцистеина. Доказано, что коферментные функции ТГФК непосредственно связаны с переносом одноуглеродных групп, первичными источниками которых в организме являются β-углеродный атом серина, α-углеродный атом глицина, углерод метальных групп метионина, холина, 2-й углеродный атом индольного кольца триптофана, 2-й углеродный атом имидазольного кольца гистидина, а также формальдегид,муравьиная кислота и метанол. К настоящему времени открыто шесть одноуглеродных групп, включающихся в разнообразные биохимические превращения в составе ТГФК: формильная (—СНО), метильная (—СН3), метиленовая (—СН2—), метенильная (—СН=), оксиметильная (—СН2ОН) и формими-новая (—CH=NH). Выяснено, что присоединение этих фрагментов к ТГФК является ферментативной реакцией ковалентного связывания их с 5-м или 10-м атомом азота (или с обоими атомами вместе). Имеются данные, что производные ТГФК участвуют в переносе одно-углеродных фрагментов при биосинтезе метионина и тимина(перенос метильной группы), серина (перенос оксиметильной группы), образовании пуриновых нуклеотидов (перенос формильной группы) и т.д. Недостаточность фолиевой кислоты у человека возникает редко. Гиповитаминоз фолиевой кислоты приводит к нарушению обмена одноуглеродных фрагментов. Такое же нарушение наблюдается и при недостаточности витамина В12, использование которого связано с обменом фолиевой кислоты. Первое проявление дефицита фолиевой кислоты - мегалобластная (макроцитарная) анемия. Она характеризуется уменьшением количества эритроцитов, снижением содержания в них гемоглобина, что вызывает увеличение размера эритроцитов. Причина этих симптомов - нарушение синтеза ДНК и РНК из-за недостатка их предшественников - тимидиловой кислоты и пуриновых нуклеотидов вследствие дефицита производных Н4-фолата. Клетки кроветворной ткани быстро делятся, поэтому они в первую очередь реагируют на нарушение синтеза нуклеиновых кислот снижением скорости эритропоэза. Мегалобластная анемия возникает чаще всего в результате недостаточности фолиевой кислоты и/или витамина В12. Гомоцистин может накапливаться в крови и тканях" выделяться с мочой, вызывая гомоцистинурию. Возможной причиной является наследственное нарушение обмена гомоцистеина либо гиповитаминоз фолиевой кислоты, а также витаминов В12 и В6 32. Недостаточность фолиевой кислоты и витамина В12. Антивитамины фолиевой кислоты. Механизм действия сульфаниламидных препаратов. Недостаточность фолиевой кислоты у человека возникает редко. Гиповитаминоз фолиевой кислоты приводит к нарушению обмена одноуглеродных фрагментов. Такое же нарушение наблюдается и при недостаточности витамина В12, использование которого связано с обменом фолиевой кислоты. Первое проявление дефицита фолиевой кислоты - мегалобластная (макроцитарная) анемия. Она характеризуется уменьшением количества эритроцитов, снижением содержания в них гемоглобина, что вызывает увеличение размера эритроцитов. Причина этих симптомов - нарушение синтеза ДНК и РНК из-за недостатка их предшественников - тимидиловой кислоты и пуриновых нуклеотидов вследствие дефицита производных Н4-фолата. Клетки кроветворной ткани быстро делятся, поэтому они в первую очередь реагируют на нарушение синтеза нуклеиновых кислот снижением скорости эритропоэза. Мегалобластная анемия возникает чаще всего в результате недостаточности фолиевой кислоты и/или витамина В12. В медицинской практике (в частности, в онкологии) нашли применение некоторые синтетические аналоги (антагонисты) фолиевой кислоты. Так, метатрескат, казывает выраженное иммуносупрессивное действие даже в относительно низких дозах, не обладающих заметной гематологической токсичностью. Благодаря этому метотрексат шире, чем другие цитостатики с иммуносупрессивной активностью, применяется в качестве иммуносупрессивного препарата. Аминоптерин является наиболее активным цитостатиком-антагонистом фолиевой кислоты; отличается высокой токсичностью, вследствие чего показан лишь при тяжёлых формах псориаза. Все современные сульфаниламидные препараты сходны между собой по спектру и механизму противомикробного действия. К ним весьма чувствительны стрептококки, стафилококки, пневмококки, гонококки, менингококки, кишечная, дизентерийная, дифтерийная и сибиреязвенная палочки, а также холерные вибрионы, бруцеллы и хламидии (возбудители трахомы и др.). На микроорганизмы сульфаниламиды оказывают бактериостатическое влияние. Механизм бактериостатического действия сульфаниламидов заключается в том, что эти вещества, имея структурное сходство с пара-аминобензойной кислотой (ПАБК), оказываются ее конкурентными антагонистами. ПАБК необходима микроорганизмам для синтеза фолиевой кислоты, которая превращается в фолиниевую кислоту, участвующую в синтезе нуклеиновых кислот. Синтез нуклеиновых кислот, как известно, является основным фактором, обеспечивающим развитие и размножение любых клеток, в том числе микроорганизмов. Замещая ПАБК в процессе синтеза фолиевой кислоты, сульфаниламиды нарушают образование этой кислоты и таким образом препятствуют образованию нуклеиновых кислот, что сопровождается задержкой развития и размножения микроорганизмов . Для развития клеток организма человека также необходима фолиевая кислота. Однако в отличие от микроорганизмов клетки человека сами не синтезируют фолиевую кислоту, а поглощают ее из крови, в которую эта кислота всасывается из кишечника. Этим объясняется тот факт, что клетки человека практически нечувствительны к действию сульфаниламидов в отличие от микроорганизмов. Особенностями механизма действия сульфаниламидов объясняется также и то, что в средах с высоким содержанием ПАБК (кровь, гной) антибактериальная активность сульфаниламидов заметно снижается. Аналогичное явление наблюдается в случае применения сульфаниламидов совместно с лекарственными веществами, при распаде которых в организме выделяется ПАБК (например, с новокаином). Действие сульфаниламидов ослабляется также при совместном применении с фолиевой кислотой или с веществами, участвующими в ее синтезе (например, с метионином). 33. Обмен фенилаланина и тирозина. Все пути превращения в норме. Фенилаланин - незаменимая аминокислота, так как в клетках животных не синтезируется её бензольное кольцо. Тирозин - условно заменимая аминокислота, поскольку образуется из фенилаланина. Содержание этих аминокислот в пищевых белках (в том числе и растительных) достаточно велико. Фенилаланин и тирозин используются для синтеза многих биологически ктивных соединений. В разных тканях метаболизм этих аминокислот происходит по-разному. 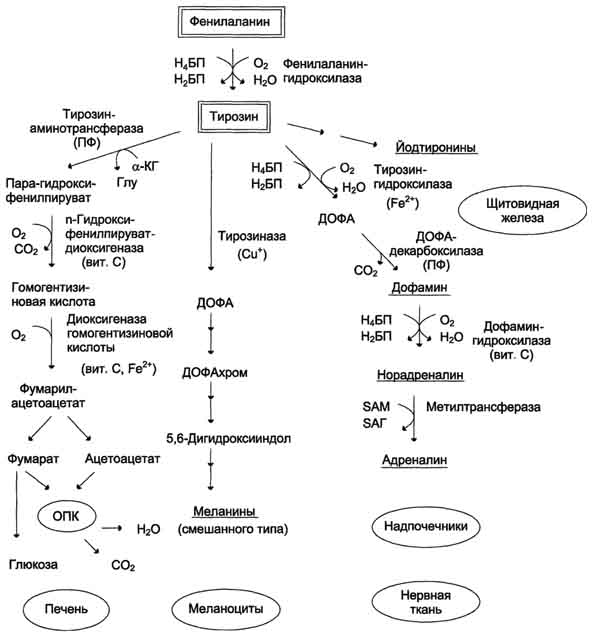 Метаболизм феиилаланина Основное количество фенилаланина расходуется по 2 путям: включается в белки; превращается в тирозин. Превращение фенилаланина в тирозин прежде всего необходимо для удаления избытка фенилаланина, так как высокие концентрации его токсичны для клеток. Образование тирозина не имеет большого значения, так как недостатка этой аминокислоты в клетках практически не бывает. Основной путь метаболизма фенилаланина начинается с его гидроксилирования, в результате чего образуется тирозин. Эта реакция катализируется специфической монооксиге-назой - фенилаланингидр(жсилазой, кофермен-том которой служит тетрагидробиоптерин (Н4БП). Активность фермента зависит также от наличия Fe2+. Реакция необратима. Н4БП в результате реакции окисляется в дигидробиоптерин (Н2БП). Регенерация последнего происходит при участии дигидроптеридинредуктазы с использованием NADPH + H+. Обмен тирозина значительно сложнее, чем обмен фенилаланина. Кроме использования в синтезе белков, тирозин в разных тканях выступает предшественником таких соединений, как катехоламины, тироксин, меланины, и ка-таболизируется до СО2 и Н2О. |