|
|
Ллкд. 1. Общие вопросы биоинформатики Объекты исследования биоинформатики. Выберите один или несколько правильных ответов
Укажите ПО\сервисы, входящие в состав BLAST на NCBI (выберите один или несколько правильных ответов)?
Blastn и blastp.
Psi-blast.
Blastx и tbastx.
Tblastn
Все вышеперечисленное.
Дана следующая матрица аминокислотных замен:
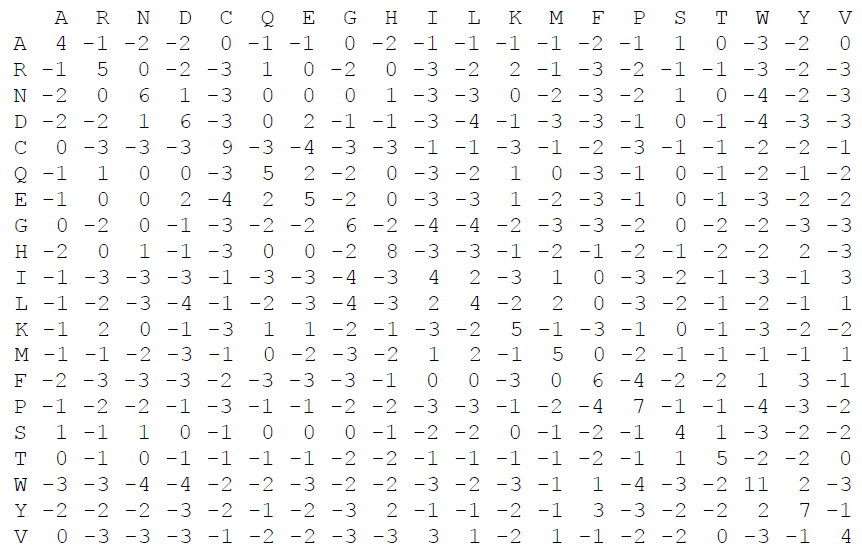
Открытие промежутка – -8. Удлинение промежутка – -1.
Какова стоимость выравнивания (выберите один правильный ответ):
AACDQRST
A-CD--ST
13
11
17
21
26
Дана следующая матрица аминокислотных замен:
Открытие промежутка – -8. Удлинение промежутка – -1.
Какова стоимость выравнивания (выберите один правильный ответ):
AACDQRST
A-CD-SST
13
11
17
19
10
По имеющимся последовательностям и матрице замен у Вас получилась следующая «трассировка» выравнивания:

Какое из выравниваний соответствует этой трассе (расставьте правильно гэпы) (выберите один правильный ответ):
ACA_CCTTA
CCATCCA_A
A_CACCTT__A
CC_A___TCCAA
ACA_CCTTA
CCATCC_AA
ACACCTTA
CCATCC_AA
ACACCTTA
CCATCCAA
Дано две последовательности белков со слабым, отдалённым сходством. Также Вы знаете последовательности их генов. Почему Вы можете ожидать лучшее выравнивание при использовании последовательностей белков (Выберите один правильный ответ)?
Потому что при сравнении дистантных белков по аминокислотной последовательности, с учётом избыточности генетического кода, выравнивание будет некачественным при использовании матриц PAM c высокими индексами (PAM200-PAM250).
Потому что при сравнении дистантных белков по аминокислотной последовательности, с учётом избыточности генетического кода, выравнивание будет качественным только при использовании матриц PAM c низкими индексами (PAM1-PAM80).
Это из-за альтернативного сплайсинга.
Потому что при сравнении дистантных белков по нуклеотидной последовательности кодирующих генов, с учётом избыточности генетического кода, выравнивание будет качественным только при использовании матриц BLOSUM с высоким индексом (больше, чем BLOSUM62).
Потому что при сравнении дистантных белков по нуклеотидной последовательности кодирующих генов, с учётом избыточности генетического кода, выравнивание может дать искажённые, сильно заниженные результаты. Кроме того, матрицы BLOSUM чувствительнее к таким случаям, чем PAM-матрицы.
Когда мы анализируем выравнивание аминокислотных последовательностей, мы обращаем внимание не только на совпадение\несовпадение символов, а ещё и на похожесть представляемых ими аминокислот. Какая биологическая логика за этим стоит (Выберите один правильный ответ)?
Похожие аминокислоты скорее всего не приведут к серьёзным изменениям в структуре белка\активного центра и скорее всего функция белка сохранится.
Случайные мутации происходят таким образом, что после трансляции аминокислоты заменяются на похожие.
Синонимичные замены в ДНК всегда приводят к замене одной аминокислоты на похожую.
Похожесть аминокислот при мутагенезе – суть эволюции макромолекул в природе.
Мутагенез всегда протекает в паре похожих аминокислот, что в свою очередь не позволяет данному белку «вымываться» из эволюции.
В чём различие между итеративным BLAST (psi-blast) и обычным поиском BLAST (Выберите правильный ответ)?
BLAST проводит поиск по аминокислотным последовательностям, psi-BLAST – по нуклеотидным.
BLAST и его аналог psi-BLAST – суть одно и то же, но BLAST на серверах NCBI, а psi-BLAST – на EMBL-EBI.
Psi-BLAST проводит поиск значительно быстрее BLAST за счёт механизмов искусственного интеллекта.
BLAST – это бесплатная (и потому медленная) версия программы поиска гомологов. Psi-BLAST – платная версия, в которой отсутствуют ограничения по скорости.
BLAST при помощи обычной процедуры выравнивания находит в БД гомологи, psi-BLAST использует при поиске построение профиля MSA, что облегчает поиск дистантных гомологов.
Дано две случайно сгенерированных нуклеотидных последовательности. Ожидалось, что в их выравнивании с промежутками будет 25% идентичных нуклеотидов в столбцах. Однако при проверке их оказалось больше, чем 25%. Почему (Выберите один правильный ответ)?
Причина в длине последовательностей – слишком короткие.
Причина в параметрах алгоритма выравнивания и в расставленных гэпах.
Причина в длине последовательностей – слишком длинные.
Причина в алгоритме рандомизации – компьютер генерирует псевдослучайную последовательность.
Мне дали не случайные последовательности, а сказали, что они были сгенерированы случайно.
Есть выравнивание двух аминокислотных последовательностей с 20% идентичности. Как проверить достоверность выравнивания (Выберите один правильный ответ)?
Провести бутстрэп-тест и посчитать alignment score.
Посчитать E-value.
Вычислить p-value.
Сравнить выравнивание структур этих белков.
Достоверность для парного выравнивания не проверяется. Она есть только для множественного выравнивания.
При множественном выравнивании последовательностей нужно максимизировать оценку (score).
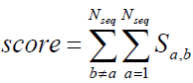
Что такая запись обозначает словами (Выберите один правильный ответ)?
Первая (справа) сумма – это score парного выравнивания последовательности a и последовательности b. Вторая (слева) сумма – score выравнивания b с последовательностью, отличающейся от a. В итоге – общая оценка MSA.
Первая (справа) сумма – это score парных выравниваний первой последовательности в наборе со всеми остальными. Вторая (слева) сумма – суммирование оценок из первой суммы по всем b. В итоге – общая оценка MSA.
Первая (справа) сумма – это score парных выравниваний последовательности b с любой последовательностью из диапазона 1<=a<=N. Вторая (слева) сумма – это просто правила записи для дальнейшего построения алгоритма. В итоге – общая оценка MSA.
Первая (справа) сумма – это score парных выравниваний последовательности b со всеми остальными. Вторая (слева) сумма – суммирование оценок из первой суммы по всем b. В итоге – общая оценка MSA.
Первая (справа) сумма – это score парных выравниваний последовательности b со всеми остальными. Вторая (слева) сумма – суммирование оценок из первой суммы по всем a. В итоге – общая оценка MSA.
При MSA один из этапов – построение guide tree. Каким образом узлы объединяются вместе (Выберите один правильный ответ)?
При помощи динамического программирования.
При помощи distance based алгоритмов кластеризации (UPGMA или Neighbor joining).
При помощи алгоритмов, основанных на похожести/оценках вероятности (Maximal parsimony, maximum likelihood).
При помощи упорядочивания входного файла последовательностей.
Есть три последовательности A, B, C. Последовательности B и C некоторым образом попарно выравниваются с А. Получить хорошее парное выравнивание В и С не удаётся. Почему (Выберите один или несколько правильных ответ)?
Производится выравнивание по нуклеотидным последовательностям (следует провести выравнивание по аминокислотным последовательностям).
В и С – последовательности разных доменов. А – последовательность многодоменного белка, в составе которой есть В и С.
Используется матрица замен, не соответствующая поставленной задаче или для сравнения близкородственных последовательностей.
При небольшом проценте идентичности (<40%) транзитивность в выравнивании может нарушаться.
А и С – последовательности разных доменов. В – последовательность многодоменного белка, в составе которой есть А и С.
После построения MSA была подсчитана вариабельность\консервативность аминокислот (S) как функция от позиции аминокислоты в последовательности (см.рисунок). Аминокислоты 37 и 43 показывают выраженную консервативность. Почему эти аминокислоты могут быть важны (Выберите один правильный ответ)?

Потому что индекс изменчивости к них меньше 0,5 (см. рисунок).
Потому что порядковый номер этих аминокислот говорит о том, что они ближе к N-концу белка, а значит, транслируются раньше остальных.
Потому что факт эволюционирования и «фиксации» в поколениях и видах определённых позиций говорит о том, что эти аминокислоты обеспечивают выполнение жизненно важной функции.
Потому что скорее всего они находятся на поверхности белка и участвуют в межбелковом взаимодействии.
Потому что из литературных данных все аминокислоты с индексом изменчивости меньше 0,5 – важные аминокислоты.
Был построен MSA для 500 последовательностей фермента тирозин киназа. Оказалось, что очень мало сайтов демонстрирует эволюционную консервативность. Что нужно сделать, чтобы в MSA этих сайтов стало больше? (Выберите один или несколько правильных ответов)?
При помощи psi-BLAST и поиску по профилю добавить в MSA дистантные гомологи, возможно – иные ферменты.
Провести попарные выравнивания для 500 последовательностей. Анализ сайтов проводить по парным выравниваниям.
Построить MSA без guide tree.
Убрать из списка последовательностей дистантные гомологи.
Изменить параметры выравнивания и построения guide tree (матрицы замен для дистантных гомологов, ослабить штрафы за промежутки).
Считается, что аминокислотные последовательности изменяются (мутируют\эволюционируют) в несколько раз быстрее, чем структуры белков. Почему (Выберите один или несколько правильных ответов)?
Пространство возможных структур заметно меньше, чем пространство возможных последовательностей в силу физических законов.
Известно, что одна и та же третичная структура может соответствовать негомологичным, с низкой идентичностью первичным структурам.
Очень ограниченное количество мутаций в определённых местах последовательности приводит к значительной перестройке третичной структуры белка.
Изменчивость третичной структуры детерминирована не только мутациями в последовательности, но и клеточной локализацией белка.
Изменчивость третичной структуры детерминирована не только мутациями в последовательности, но и фазой клеточного цикла.
Нужно построить множественное выравнивание для n последовательностей. Сколько парных выравниваний нужно построить для этого (Выберите один правильный ответ)?
n^2/2.
n*((n-1)/2).
2^(n-1).
n*(n-1)/2.
(n-1)^2.
Тема 3. Биоинформатика и структурная биология белков
Вторичные структуры белков. Выберите один или несколько правильных ответов:
Белковые домены
Повороты
Альфа-спирали
«Цинковые пальцы» (Zinc fingers) у факторов транскрипции
Бета-листы
Что является группой аминокислот. Выберите один или несколько правильных ответов:
Неполярные.
Смешанные
Содержащие ароматический радикал
Пуриновые
Заряженные положительно при pH=7
Третичная структура – это (выберите один правильный ответ):
пространственное строение (включая конформацию) всей молекулы белка или другой макромолекулы.
Пространственное строение (включая все конформации) всех боковых цепей аминокислот белка.
Векторное представление элементов вторичной структуры белка.
Формула SMILES последовательности белка.
3.4. Фолдинг белка – (выберите один правильный ответ):
Очень часто протекает при участии шаперонов.
Некоторые нейродегенеративные заболевания являются результатом нарушения фолдинга определённых белков.
Физический процесс спонтанного сворачивания полипептидной цепи в нативную пространственную структуру.
Все вышеперечисленное.
3.5 Какие существуют методы предсказания третичной структуры белка (выберите один или несколько правильных ответов):
Метод GOR.
Ab-initio метод.
Распознавание фолда.
Метод ближайшего соседа.
Моделирование по гомологии.
3.6. Качественное моделирование структуры белка по гомологии невозможно без (выберите один или несколько правильных ответов):
Процента идентичности между последовательностями исследуемого белка и шаблонного не менее 25%.
Наличия в PDB структуры шаблонного белка.
Наличия в шаблонной 3D структуре элементов вторичной структуры в достаточном количестве.
Минимизации свободной энергии белка в качестве заключительного этапа моделирования.
Моделирование структуры белка по гомологии – назовите известные методы или сервисы (выберите один или несколько правильных ответов):
Phyre2.
Swiss-Model.
HHPred.
I-Tasser.
IntFOLD.
Распознавание фолда (threading) применяется для (выберите один правильный ответ):
Предсказание связывания двух гомологичных белков.
Предсказание надструктурных элементов белка.
Предсказание элементов вторичной структуры белка.
Предсказание третичной структуры белка при высокой идентичности последовательностей.
Предсказание третичной структуры белка при невысокой идентичности последовательностей.
Моделирование структуры белка при помощи распознавания фолда – назовите известные методы или сервисы (выберите один или несколько правильных ответов):
Phyre2.
Swiss-Model.
RaptorX.
I-Tasser.
HHPred.
Классификации белков (выберите один или несколько правильных ответов):
По форме.
По количеству водородных связей.
По количеству дисульфидных мостиков.
По составу.
По функциям.
База данных Pfam – это (выберите один правильный ответ):
База данных групп белков.
База данных аминокислот.
База данных семейств белков.
База данных прионных белков.
База данных бесструктурных белков.
База данных SCOP и SCOP2 – это (выберите один правильный ответ):
База данных групп белков.
Структурная классификация белков.
База данных семейств белков.
База данных факторов транскрипции.
Классификация бесструктурных белков.
CATH – это (выберите один правильный ответ):
База данных групп белков.
Классификация бесструктурных белков.
База данных семейств белков.
База данных факторов транскрипции.
Структурная классификация доменов белков.
Назовите методы экспериментального определения структуры белка (Выберите один или несколько правильных ответов):
Рентгеноструктурный анализ.
Ядерно-магнитный резонанс.
Электронная микроскопия.
Исследование гомологии.
Выравнивание структур.
Назовите методы/сервисы оценки качества полученной в результате эксперимента или моделирования структуры белка (Выберите один или несколько правильных ответов):
TCoffee.
ProCheck.
What_If.
ClustalW.
MolProbity.
Метод GOR и Chou-Fasman применяются для (Выберите один правильный ответ):
Предсказания третичной структуры по последовательности белка.
Выравнивания третичных структур белков.
Предсказания вторичных структур по последовательности белка.
Предсказания первичной структуры белка по его вторичной структуре.
Планирования X-Ray эксперимента при помощи CAOS или Chem-Ray.
Какой тип представления следует выбрать в любой программе для визуализации белков, чтобы увидеть и проанализировать элементы вторичной структуры (Выберите один или несколько правильных ответов)?
Ribbons.
Secondary.
Balls and sticks.
Pretty lines.
Cartoons.
Дан PDB файл, в котором структура была исследована при помощи рентгеноструктурного анализа с разрешением 1.5 Å. Между тем в главной цепи расстояния C-N равны 1.32 Å. Объясните этот феномен (Выберите один правильный ответ)?
Чтобы локализовать короткие ковалентные связи в главной цепи белка используются повторяющиеся серии рентгеноструктурных экспериментов.
Для распознавания положения такой ковалентной связи в главной и боковых цепей используются метки изотопа углерода С13.
На первом этапе X-Ray исследования связи C-N имеют длину, равную или превышающую разрешение установки. На втором этапе, зная длину всей молекулы, связи C-N масштабируются для соответствия известной длине белка.
Достаточно точно определить положение первого атома N а каждой аминокислоте. Затем программа сама достроит правильно главную и боковые цепи всего белка.
Известно, что разрешение в 2Å приводит к погрешности в определении координат в 0,2Å. Поскольку радиус Ван-дер-Ваальса атома углерода – 3,4-3,7 Å, то алгоритмы постобработки могут определить центры атомов с большой точностью при указанном разрешении X-Ray.
Нужно сравнить структуры двух белков по формуле стандартного отклонения (см. рисунок). Nres – количество аминокислот, raiи rbi – векторы координат Сa атомов i-аминокислоты белка A и белка B соответственно.

Какую процедуру нужно выполнить над структурами белков прежде, чем применять эту формулу для их сравнения (выберите один правильный ответ)?
Процедуру выравнивания последовательностей.
Процедуру молекулярного докинга.
Процедуру моделирования по гомологии.
Запустить процедуру молекулярной динамики или минимизировать энергии обоих белков.
Процедуру структурного выравнивания.
Тема 4. Молекулярный докинг и скоринг
Докинг – это (выберите один правильный ответ):
Метод молекулярного моделирования, целью которого является конструирование нового органического соединения.
Метод моделирования белковой структуры, основанный на распознавании фолда.
Метод математического моделирования всасывания, распределения и выведения лекарственного средства и его метаболитов из организма.
Метод молекулярного моделирования, целью которого является нахождение ориентации одной молекулы по отношению к другой при образовании комплекса.
Одна из разновидностей структурного выравнивания.
Скоринг – это (выберите один правильный ответ):
Метод улучшения положения лиганда в активном центре при помощи молекулярно-динамической симуляции.
Оценка качества, вероятностей положений молекул в молекулярном комплексе, выраженная как сила нековалентных взаимодействий.
Метод построения больших библиотек органических молекул.
Оценка различий в структурах белков, выраженная в виде среднеквадратичного отклонения (в ангстремах).
Одна из разновидностей структурного выравнивания, основанная на вычислении TM-score.
Программное обеспечение для докинга и скоринга (Выберите один или несколько правильных ответов):
SwissPDB-viewer.
Glide.
AutoDock
GROMACS.
GOLD.
Вопросы, для ответа на которые производится молекулярный докинг (Выберите один или несколько правильных ответов):
Связываются данные молекулы или нет.
Насколько качественно и быстро применяемое ПО может предсказать связывание данных молекул.
Насколько сильно данные молекулы связываются.
Как выглядит комплекс из двух данных молекул.
Подходит ли применяемое поле сил для данного растворителя и интересующих молекул.
Перечислите некоторые трудности молекулярного докинга (Выберите один или несколько правильных ответов):
Выбор правильной функции оценки (скоринга)
Некоторые научные статьи утверждают, что данные молекулы не связываются.
Имеющаяся видеокарта не поддерживает отображение сложной графики, которая требуется для отображения всех структур.
Определение правильной структуры сайта связывания.
В коллективе отсутствует химик-синтетик, способный синтезировать требуемый лиганд.
Жёсткий докинг – это (Выберите один правильный ответ):
В процессе докинга две молекулы жёстко связываются между собой.
В процессе докинга у исследуемых молекул длины ковалентных связей и планарные углы не меняются.
В процессе докинга у исследуемых молекул торсионные углы меняются в ограниченном диапазоне (около 50 гр).
В процессе докинга у исследуемых молекул длины ковалентных связей, планарные и торсионные углы не изменяются
В процессе докинга имеющиеся молекулы растворителя остаются неподвижны.
Ситуации, при которой жёсткий докинг может дать относительно адекватные результаты (Выберите один или несколько правильных ответов):
Оценка энергии уже имеющегося комплекса.
Докинг лигандов с цепями ненасыщенных жирных кислот.
Докинг лигандов с атомами металлов.
Докинг пептидов и ДНК.
Докинг небольшого лиганда, часто с ароматическим кольцом.
Гибкий докинг – это (Выберите один или несколько правильных ответов):.
Процедура докинга, которая допускает изгиб молекул в горизонтальной плоскости.
Процедура докинга, которая допускает конформационные изменения молекул.
Процедура докинга, которая не допускает изменения торсионных углов.
Процедура «подгонки» лиганда под кривизну поверхности рецептора.
Специальная процедура докинга, разработанная для аминокислот.
Тема 5. Филогения и филогенетические деревья
Алгоритмы построения филогенетических деревьев (выберите один или несколько правильных ответов):
CombiGlide.
UPGMA.
Neighbor Joining.
Алгоритм Дейкстры.
Алгоритм максимальной бережливости.
Свойства подхода UPGMA (выберите один или несколько правильных ответов):
Базируется на гипотезе о верности «молекулярных часов».
Расстояние всех OTU от корня одинаково.
c. На последнем этапе подход использует полный перебор всех возможных филогенетических деревьев для повышения точности.
d. Для его работы матрица расстояний не нужна.
e. Концептуально схож с генетическими алгоритмами.
Свойства алгоритма Neighbor Joining (выберите один или несколько правильных ответов):
Применим только к последовательностям белков.
Результат – 3 различных топологии филогенетического дерева.
c. Строит неукоренённые филогенетические деревья.
d. Медленный алгоритм, неприменим к большим наборам данных.
e. Не привязан к гипотезе «молекулярных часов».
Свойства алгоритма максимальной бережливости (Maximum Parsimony) (выберите один или несколько правильных ответов):
Применим только к последовательностям ДНК.
b. Строит филогенетическое дерево, объясняющее экспериментальные данные наименьшим числом эволюционных событий.
c. Базируется на матрице расстояний.
d. Медленный алгоритм, неприменим к большим наборам данных.
Молекулярная вычислительная филогенетика – это:
Способ сбора и классификации морфологических данных.
Способ восстановления родственных связей, реконструирование эволюционных событий и построение филогенетических деревьев.
Способ установления родственных связей между членами одной семьи.
Способ сравнения последовательностей ДНК, в основе которого лежат алгоритмы машинного обучения.
5.6. Гипотеза «молекулярных часов» утверждает, что (выберите один правильный ответ):
Количество значимых замен в одном гене в разных ветвях (видах) ускоряется со временем – антропогенные факторы.
Количество значимых замен в одном гене в разных ветвях (видах) имеет синусоидальную зависимость от времени – геологические периоды\эры.
Время расхождения видов от общего предка можно точно определить при помощи радиоизотопного анализа.
Количество значимых замен в одном гене в разных ветвях (видах) имеет линейную зависимость от времени.
5.7. Клада – это (выберите один правильный ответ):
Группа последовательностей (OTU) и их общий предок.
Группа последовательностей (OTU) и корень филогенетического дерева.
Все узлы филогенетического дерева.
Все узлы и корень филогенетического дерева.
Представители всех таксонов филогенетического дерева с их общими предками.
Операционная таксономическая единица (OTU) – это (выберите один правильный ответ):
Внутренний узел филогенетического дерева.
Мера длины ветви филогенетического дерева.
Характеристика того или иного таксона в филогенетическом дереве.
Уровень детализации в филогении, обычно – последовательность вида, располагающаяся в листе филогенетического дерева.
Ветвь филогенетического дерева – это (выберите один правильный ответ):
Упрощённая топология филогенетического дерева.
Форма записи последовательности общих для предка и потомка генов.
Определяет взаимоотношение между таксонами, предками и потомками. Ребро графа филогенетического дерева.
Показывает направление эволюции в неукоренённых филогенетических деревьях.
5.10. Лист филогенетического дерева – это (выберите один правильный ответ):
Реальный современный объект. Последовательность, соответствующая внешней вершине графа.
Узел филогенетического дерева у лиственничных растений.
Последовательность, соответствующая внутреннему узлу филогенетического дерева.
Узел филогенетического дерева с исходящими из него тремя ветвями.
Корень филогенетического дерева – это (выберите один правильный ответ):
Название листа филогенетического дерева, от которого отходят несколько ветвей.
Узел филогенетического дерева, от которого отходит самая толстая ветвь к ближайшему листу.
Общий предок для всех изучаемых последовательностей в филогенетическом дереве.
Множество узлов филогенетического дерева, расстояние между которыми минимально (суперузел).
Множество ветвей филогенетического дерева одинаковой длины, расстояние между которыми минимально (суперветвь).
Ультраметрическое филогенетическое дерево – это (выберите один правильный ответ):
Дерево, в котором все расстояния от корня до листьев неодинаковы.
Дерево, в котором встречаются как равные, так и неравные расстояния от корня до листьев.
Филогенетическое дерево без корня (неукоренённое дерево).
Филогенетическое дерево, которое имеет смысл только для прокариот.
Дерево, в котором все расстояния от корня до листьев равны.
Неультраметрическое филогенетическое дерево – это (выберите один правильный ответ):
Дерево, в котором все расстояния от корня до листьев неодинаковы.
Дерево, в котором встречаются как равные, так и неравные расстояния от корня до листьев.
Филогенетическое дерево без корня (неукоренённое дерево).
Филогенетическое дерево, которое имеет смысл только для прокариот.
Дерево, в котором все расстояния от корня до листьев равны.
Ортологи – это (выберите один правильный ответ):
Гомологичные последовательности, образовавшиеся в результате дупликации.
Гомологичные последовательности, образовавшиеся в результате события видообразования.
Гомологичные последовательности, образовавшиеся в результате горизонтального переноса генов
Негомологичные последовательности, кодирующие структурно сходные белки.
Паралоги – это (выберите один правильный ответ):
Гомологичные последовательности, образовавшиеся в результате события видообразования.
Гомологичные последовательности, образовавшиеся в результате горизонтального переноса генов.
Гомологичные гены, образовавшиеся в результате события дупликации.
Негомологичные последовательности, кодирующие структурно сходные или функционально связанные белки.
Гомологи – это (выберите один правильный ответ):
Последовательности гомологичны, если две и более программ для парного выравнивания дают их идентичность более 65%.
Последовательности генов гомологичны, если продукты этих генов (белки) имеют схожую клеточную локализацию.
Гомологи – это пара из гена и соответствующего ему белка.
Последовательности гомологичны если они произошли от одной общей для них последовательности.
Ксенологи – это (выберите один правильный ответ):
Гены ксенобактерий, применяемых для очистки почв, водоёмов от нефтепродуктов.
Гомологичные последовательности, возникшие при горизонтальном переносе генов.
Специальные белки, применяемые при производстве ксеноновых ламп.
Гены, кодирующие редкие и очень редкие белки в человеческом организме.
Таксон – это (выберите один правильный ответ):
Группа особей с общими признаками, дающая в ряду поколений плодовитое потомство.
Группа из нескольких белков, отвечающих за двигательную активность.
Группа из участков генома с регуляторными функциями.
d. Группа в классификации, состоящая из дискретных объектов, объединяемых на основании общих свойств и признаков.
Какой тест может быть проведен на филогенетическом дереве, построенном по NJ алгоритму, чтобы высказать предположение о его корректности (выберите один правильный ответ):
Тест на похожесть.
Бутстрэп-тест.
Тест на аддитивность.
Тест на похожесть.
Тест на соответствие.
Свойство аддитивности филогенетического дерева. Его определение (выберите один или несколько правильных ответов):
Для любых трёх OTU два расстояния между ними равны и не меньше третьего.
Для любых четырёх OTU сумма любых двух расстояний равна и больше третьей суммы
Бутстрэп-тест.
Для любых OTU расстояние от OTU до корня одинаково.
Для любых четырёх OTU сумма любых двух расстояний равна и меньше третьей суммы.
Что может сказать об эволюционном процессе организмов свойство ультраметричности построенного для них дерева (при условии его истинности. Выберите один правильный ответ)?
Длина ветвей – суть расстояния между последовательностями. Эволюция протекала в резко меняющейся среде.
Длина ветвей – суть время эволюции. Эволюция протекала в относительно монотонной среде.
Геномы организмов эволюционировали в том числе посредством HGT, что опосредовано видно из длины ветвей дерева.
В построенном дереве преобладают гены-ортологи.
В построенном дереве преобладают гены-паралоги.
Почему динамическое программирование (квадратический алгоритм, сложность L^n) используется для парного выравнивания и практически не применяется для выравнивания нескольких последовательностей (MSA) (Выберите один правильный ответ)?
Потому что это слишком неточно. Для 20-и последовательностей длины 200 букв вероятность найти верное выравнивание равна
1,04e-46.
Потому что не написано соответствующее программное обеспечение.
Потому что для этого нужны специализированные матрицы замен, которые в настоящее время только разрабатываются.
Потому что это слишком долго. Для 20-и последовательностей длины 200 букв получаем 200^20 (1,048576e+46 вариантов).
Потому что нужно слишком большой объём оперативной памяти компьютера для загрузки многомерной матрицы последовательностей.
Дана следующая строка: ((raccoon, bear),((sea_lion,seal),((monkey,cat), weasel)),dog). Как называется этот формат записи и что он описывает (Выберите один правильный ответ)?
Формат Newick. Описывает построчное расположение последовательностей в MSA.
Формат multi-FASTA. Описывает последовательности в fasta-файле.
Формат GenBank. В таком формате осуществляет выгрузка последовательностей из базы данных GenBank.
Формат Newick. Описывает топологию филотенетических деревьев.
Формат ClustalW. Применяется для описания результатов MSA.
Дано филогенетическое дерево. Какая строка описывает эту топологию (Выберите один правильный ответ)?

((x2,x6),((x7,(x4,(x1,x3))),(x5,x8)).
((x2,x6),((x7,(x4,(x1,x3))),(x5,x8))).
((x2,x6),((x7,(x4,(x1,x3))),(x5,x9))).
((x7,(x4,(x1,x3),((x2,x6))),(x5,x8))).
((x2,x6),(x5,x8)),((x7,(x4,(x1,x3)))).
((x2,x6),(х7,x5,x8),(x4,x1,x3)).
|
|
|
 Скачать 0.76 Mb.
Скачать 0.76 Mb.