колоквиум №2. 1. Предмет, задачи, методы генетики. История развития генетики. Роль отечественных ученых (Н. К. Кольцов, А. С. Серебровский, С. С. Четвериков) в развитии генетики
 Скачать 274.6 Kb. Скачать 274.6 Kb.
|

|
14..Рибосомный цикл синтеза белка (инициация, элонгация, терминация). Посттрансляционные преобразования белков. Рибосомный цикл синтеза белка. Процесс взаимодействия мРНК и тРНК, обеспечивающий трансляцию информации с языка нуклеотидов на язык аминокислот, осуществляется на рибосомах. Последние представляют собой сложные комплексы рРНК и разнообразных белков, в которых первые образуют каркас. Рибосомные РНК являются не только структурным компонентом рибосом, но и обеспечивают связывание их с определенной нуклеотидной последовательностью мРНК. Этим устанавливаются начало и рамка считывания при образовании пептидной цепи. Кроме того, они обеспечивают взаимодействие рибосомы и тРНК. Многочисленные белки, входящие в состав рибосом наряду с рРНК, выполняют как структурную, так и ферментативную роль. Рибосомы про - и эукариот очень сходны по структуре и функциям. Они состоят из двух субчастиц: большой и малой. У эукариот малая субчастица образована одной молекулой рРНК и 33 молекулами разных белков. Большая субчастица объединяет три молекулы рРНК и около 40 белков. Прокариотические рибосомы и рибосомы митохондрий и пластид содержат меньше компонентов. В рибосомах имеется две бороздки. Одна из них удерживает растущую полипептидную цепь, другая — мРНК. Кроме того, в рибосомах выделяют два участка, связывающих тРНК. В аминоацильном, А-участке размещается аминоацил-тРНК, несущая определенную аминокислоту. В пептидильном, П-участке располагается обычно тРНК, которая нагружена цепочкой аминокислот, соединенных пептидными связями. Образование А- и П-участков обеспечивается обеими субчастицами рибосомы. В каждый момент рибосома экранирует сегмент мРНК протяженностью около 30 нуклеотидов. При этом обеспечивается взаимодействие только двух тРНК с двумя расположенными рядом кодонами мРНК (рис. 3.31). Трансляция информации на «язык» аминокислот выражается в постепенном наращивании пептидной цепи в соответствии с инструкцией, заключенной в мРНК. Этот процесс протекает на рибосомах, которые обеспечивают последовательность расшифровки информации с помощью тРНК. В ходе трансляции можно выделить три фазы: инициацию, элонгацию и терминацию синтеза пептидной цепи. 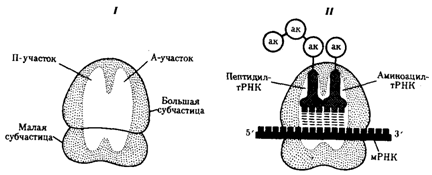 Фаза инициации, или начало синтеза пептида, заключается в объединении двух находящихся до этого порознь в цитоплазме субчастиц рибосомы на определенном участке мРНК и присоединении к ней первой аминоацил-тРНК. Этим задается также рамка считывания информации, заключенной в мРНК (рис. 3.32). В молекуле любой мРНК вблизи ее 5'-конца имеется участок, комплементарный рРНК малой субчастицы рибосомы и специфически узнаваемый ею. Рядом с ним располагается инициирующий стартовый кодон АУТ, шифрующий аминокислоту метионин. Малая субчастица рибосомы соединяется с мРНК таким образом, что стартовый кодон АУТ располагается в области, соответствующей П-участку. При этом только инициирующая тРНК, несущая метионин, способна занять место в недостроенном П-участке малой субчастицы и комплементарно соединиться со стартовым кодоном. После описанного события происходит объединение большой и малой субчастиц рибосомы с образованием ее пептидильного и аминоацильного участков (рис. 3.32). К концу фазы инициации П-участок занят аминоацил-тРНК, связанной с метионином, тогда как в А-участке рибосомы располагается следующий за стартовым кодон. Описанные процессы инициации трансляции катализируются особыми белками — факторами инициации, которые подвижно связаны с малой субчастицей рибосомы. По завершении фазы инициации и образования комплекса рибосома — мРНК — инициирующая аминоацил-тРНК эти факторы отделяются от рибосомы. Фаза элонгации, или удлинения пептида, включает в себя все реакции от момента образования первой пептидной связи до присоединения последней аминокислоты. Она представляет собой циклически повторяющиеся события, при которых происходит специфическое узнавание аминоацил-тРНК очередного кодона, находящегося в А-участке, комплементарное взаимодействие между антикодоном и кодоном. Благодаря особенностям трехмерной организации тРНК. (см. разд. 3.4.3.1) при соединении ее антикодона с кодоном мРНК. транспортируемая ею аминокислота располагается в А-участке, поблизости от ранее включенной аминокислоты, находящейся в П-участке. Между двумя аминокислотами образуется пептидная связь, катализуемая особыми белками, входящими в состав рибосомы. В результате предыдущая аминокислота теряет связь со своей тРНК и присоединяется к аминоацил-тРНК, расположенной в А-участке. Находящаяся в этот момент в П-участке тРНК высвобождается и уходит в цитоплазму (рис. 3.33). Перемещение тРНК, нагруженной пептидной цепочкой, из А-участка в П-участок сопровождается продвижением рибосомы по мРНК на шаг, соответствующий одному кодону. Теперь следующий кодон приходит в контакт с А-участком, где он будет специфически «опознан» соответствующей аминоацил-тРНК, которая разместит здесь свою аминокислоту. Такая последовательность событий повторяется до тех пор, пока в А-участок рибосомы не поступит кодон-терминатор, для которого не существует соответствующей тРНК. Сборка пептидной цепи осуществляется с достаточно большой скоростью, зависящей от температуры. У бактерий при 37 °С она выражается в добавлении к подипептиду от 12 до 17 аминокислот в 1 с. В эукариотических клетках эта скорость ниже и выражается в добавлении двух аминокислот в 1 с. Фаза терминации, или завершения синтеза полипептида, связана с узнаванием специфическим рибосомным белком одного из терминирующих кодонов (УАА, УАГ или У ГА), когда тот входит в зону А-участка рибосомы. При этом к последней аминокислоте в пептидной цепи присоединяется вода, и ее карбоксильный конец отделяется от тРНК. В результате завершенная пептидная цепь теряет связь с рибосомой, которая распадается на две субчастицы (рис. 3.34). Посттрансляционные преобразования белков. Синтезированные в ходе трансляции пептидные цепи на основе своей первичной структуры приобретают вторичную и третичную, а многие—и четвертичную организацию, образуемую несколькими пептидными цепями. В зависимости от функций, выполняемых белками, их аминокислотные последовательности могут претерпевать различные преобразования, формируя функционально активные молекулы белка. Многие мембранные белки синтезируются в виде пре-белков, имеющих на N-конце лидерную последовательность, которая обеспечивает him узнавание мембраны. Эта последовательность отщепляется при созревании и встраивании белка в мембрану. Секреторные белки также имеют на N-конце лидерную последовательность, которая обеспечивает их транспорт через мембрану. Некоторые белки сразу после трансляции несут дополнительные аминокислотные про-последовательности, определяющие стабильность предшественников активных белков. При созревании белка они удаляются, обеспечивая переход неактивного пробелка в активный белок. Например, инсулин вначале синтезируется как пре-проинсулин. Во время секреции пре-последовательность отщепляется, а затем проинсулин подвергается модификации, при которой из него удаляется часть цепи и он превращается в зрелый инсулин. I — РНК-полимераза связывается с ДНК и начинает синтезировать мРНК в направлении 5' → 3'; II — по мере продвижения РНК-полимеразы к 5'-концу мРНК прикрепляются рибосомы, начинающие синтез белка; III — группа рибосом следует за РНК-полимеразой, на 5'-конце мРНК начинается ее деградация; IV —процесс деградации протекает медленнее, чем транскрипция и трансляция; V — после окончания транскрипции мРНК освобождается от ДНК, на ней продолжается трансляция и деградация на 5'-конце Формируя третичную и четвертичную организацию в ходе посттрансляционных преобразований, белки приобретают способность активно функционировать, включаясь в определенные клеточные структуры и осуществляя ферментативные и другие функции. Рассмотренные особенности реализации генетической информации в про - и эукариотических клетках обнаруживают принципиальное сходство этих процессов. Следовательно, механизм экспрессии генов, связанный с транскрипцией и последующей трансляцией информации, которая зашифрована с помощью биологического кода, сложился в целом еще до того, как были сформированы эти два типа клеточной организации. Дивергентная эволюция геномов про - и эукариот привела к возникновению различий в организации их наследственного материала, что не могло не отразиться и на механизмах его экспресии. Постоянное совершенствование наших знаний об организации и функционировании материала наследственности и изменчивости обусловливает эволюцию представлений о гене как функциональной единице этого материала. 15 Взаимосвязь между геном и признаком. Пример. Гипотеза «один ген - один фермент», ее современная трактовка. Открытия экзон-интронной организации эукариотических генов и возможности альтернативного сплайсинга показали, что одна и та же нуклеотидная последовательность первичного транскрипта может обеспечить синтез нескольких полипептидных цепей с разными функциями или их модифицированных аналогов. Например, в митохондриях дрожжей имеется ген box (или cob), кодирующий дыхательный фермент цитохром b. Он может существовать в двух формах (рис. 3.42). «Длинный» ген, состоящий из 6400 п. н., имеет 6 экзонов общей протяженностью 1155 п. н. и 5 интронов. Короткая форма гена состоит из 3300 п. н. и имеет 2 интрона. Она фактически представляет собой лишенный первых трех интронов «длинный» ген. Обе формы гена одинаково хорошо экспрессируются. После удаления первого интрона «длинного» гена box на основе объединенной нуклеотидной последовательности двух первых экзонов и части нуклеотидов второго интрона образуется матрица для самостоятельного белка — РНК-матуразы (рис. 3.43). Функцией РНК-матуразы является обеспечение следующего этапа сплайсинга — удаление второго интрона из первичного транскрипта и в конечном счете образование матрицы для цитохрома b. Другим примером может служить изменение схемы сплайсинга первичного транскрипта, кодирующего структуру молекул антител в лимфоцитах. Мембранная форма антител имеет на С-конце длинный «хвост» аминокислот, который обеспечивает фиксацию белка на мембране. У секретируемой формы антител такого хвоста нет, что объясняется удалением в ходе сплайсинга из первичного транскрипта кодирующих этот участок нуклеотидов. У вирусов и бактерий описана ситуация, когда один ген может одновременно являться частью другого гена или некоторая нуклеотидная последовательность ДНК может быть составной частью двух разных перекрывающихся генов. Например, на физической карте генома фага ФХ174 (рис. 3.44) видно, что последовательность гена В располагается внутри гена А, а ген Е является частью последовательности гена D. Этой особенностью организации генома фага удалось объяснить существующее несоответствие между относительно небольшим его размером (он состоит из 5386 нуклеотидов) и числом аминокислотных остатков во всех синтезируемых белках, которое превышает теоретически допустимое при данной емкости генома. Возможность сборки разных пептидных цепей на мРНК, синтезированной с перекрывающихся генов (А и В или Е и D), обеспечивается наличием внутри этой мРНК участков связывания с рибосомами. Это позволяет начать трансляцию другого пептида с новой точки отсчета. Нуклеотидная последовательность гена В является одновременно частью гена А, а ген Е составляет часть гена D В геноме фага λ были также обнаружены перекрывающиеся гены, транслируемые как со сдвигом рамки, так и в той же рамке считывания. Предполагается также возможность транскрибирования двух разных мРНК с обеих комплементарных цепей одного участка ДНК. Это требует наличия промоторных областей, .определяющих движение РНК-полимеразы в разных направлениях вдоль молекулы ДНК. Описанные ситуации, свидетельствующие о допустимости считывания разной информации с одной и той же последовательности ДНК, позволяют предположить, что перекрывающиеся гены представляют собой довольно распространенный элемент организации генома вирусов и, возможно, прокариот. У эукариот прерывистость генов также обеспечивает возможность синтеза разнообразных пептидов на основе одной и той же последовательности ДНК. Имея в виду все сказанное, необходимо внести поправку в определение гена. Очевидно, нельзя больше говорить о гене как о непрерывной последовательности ДНК, однозначно кодирующей определенный белок. По-видимому, в настоящее время наиболее приемлемой все же следует считать формулу «Один ген — один поли-пептид», хотя некоторые авторы предлагают ее переиначить: «Один полипептид — один ген». Во всяком случае, под термином ген надо понимать функциональную единицу наследственного материала, по химической природе являющуюся полинуклеотидом и определяющую возможность синтеза полипептидной цепи, тРНК или рРНК. Один ген один фермент. В 1940 г Дж. Бидл и Эдвард Татум использовали новый подход для изучения того, как гены обеспечивают метаболизм у более удобного объекта исследований – у микроскопического грибка Neurospora crassa.. Ими были получены мутации, у которых; отсутствовала активность того-или иного фермента метаболизма. А это приводило к тому, что мутантный гриб бьл не способен сам синтезировать определенный метаболит (например, аминокислоту лейцин) и мог жить только тогда, когда лейцин был добавлен в питательную среду. Сформулированная Дж. Бидлом и Э. Татумом теория "один ген - один фермент" - быстро получила широкое признание у генетиков, а сами они были награждены Нобелевской Премией. Методы. селекции так называемых "биохимических мутаций", приводящих к нарушениям действия ферментов, обеспечивающих разные пути метаболизма, оказались очень плодотворными не только для науки, но и для практики. Сначала они привели к возникновению генетики и селекции промышленных микроорганизмов, а потом и к микробиологической промышленности, которая использует штаммы микроорганизмов, сверх продуцирующие такие стратегически важные вещества, какантибиотики, витамины, аминокислоты и др.. В основе принципов селекции и генной инженерии штаммов сверхпродуцентов лежит представление, что "один ген кодирует один фермент". И хотя это представление отлично практике приносит многомиллионные прибыли и спасает миллионы жизней (антибиотики) - оно не является окончательным. Один ген - это не только один фермент. 16Ген как единица изменчивости. Генные мутации и их классификация. Причины и механизмы возникновения генных мутаций. Генные болезни человека. Примеры. Ген-участок ДНК, который несет в себе информацию о строении какого либо белка Свойства: 1 дискретность действия- развитие различных признаков контролируется разными генами. 2 стабильность - передается в ряду поколений в неизменном виде. 3 специфичность - каждый из генов обуславливает развитие определенного признака. 4 плейотропия - способность генов обеспечивать развитие одновременно нескольких признаков Генные мутации связаны с изменением внутренней структуры генов, что превращает одни аллели в другие. Можно выделить несколько типов генных мутаций на молекулярном уровне: - замена пар нуклеотидов - делеция - вставка нуклеотида - перестановка (инверсия) участка гена. Замена пар нуклеотидов. Точковые мутации с заменой оснований разделяют на два класса: транзиции (и трансверсии Замена пуринового основания на другое пуриновое, или одного пиримидинового на другое пиримидиновое – транзиция. Замена пуринового основания на пиримидиновое и наоборот – трансверсия. При замене нуклеотидов в структурных генах происходит изменение смысла гена – возникают миссенс-мутации. При этом одна аминокислота в полипептиде замещается другой. Фенотипическое проявление мутации зависит от положения аминокислоты в полипептиде. При замене последовательности ЦТЦ на ЦАЦ возникает серповидно-клеточная анемия. Образуется новый полипептид и гемоглобин имеет совсем другие свойства. Некоторые миссенс-мутации приводят к возникновению фермента, обладающего высокой активностью в одних условиях и средней в других условиях. Т. к. генетический код вырожден, то при замене триплетов, кодирующий одну и ту же аминокислоту, мутации не проявляются. Другой вид мутаций – нонсенс - мутации. При этих мутациях при замене одного нуклеотида другим образуются бессмысленные триплеты. Синтез полипептида прекращается и белок имеет совсем иные свойства. УАГ. УАА. УГА. бессмысленные триплеты. Делеция или вставка одного или нескольких нуклеотидов ведут за собой утрату или вставку одной или нескольких аминокислот в полипептиде. эффекта может не быть. Если происходят делеция или вставка 1 нуклеотида (или другого числа нуклеотидов не кратного 3), наблюдается сдвиг рамки считывания, при этом нарушается структура полипептида. Большинство изменений молекулярной структуры генов приводит к новым формам считывания генетической информации, которая реализуется в метаболических путях и биохимических реакциях, появляются новые свойства клеток и всего организма. В организме происходит большое количество мутаций. Они затрагивают интеллект, поведение, метаболические признаки и т. д. мутации, изменяющие видимыеморфологические признаки – видимые (мутация альбинизма). Нормальный доминантный ген превращается в рецессивный, выработка меланина прекращается, фенотипически проявляется белой окраской волос и глаз. Есть группа биохимических мутаций, которые выявляются с помощью сложных биохимических способов. Например, у человека синтезируется ряд ферментов, осуществляющих превращение лактозы в галактозу. При отсутствии фермента-лактазы происходитброжение в толстом кишечнике, газообразование и др. могут быть детская и взрослая формы. При накоплении галактозы – галактоземия, которая может привести к умственной отсталости. Мутации, нарушающие жизнь – летальные, полулетальные и сублетальные. Летальные – гибель зиготы или развившегося организма на определенной стадии эмбриогенеза – выкидыши. Полулетальные и сублетальные ослабляют жизнеспособность организма или отдельных клеток (например, брахидактилия – гомозиготы погибают). Генные болезни – это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. Болезни аминокислотного обменаСамая многочисленная группа наследственных болезней обмена веществ. Почти все они наследуются по аутосомно-рецессивному типу. Причина заболеваний — недостаточность того или иного фермента, ответственного за синтез аминокислот. К ним относится: фенилкетонурия - нарушение превращения фенилаланина в тирозин из-за резкого снижения активности фенилаланингидроксилазы; алкаптонурия - нарушение обмена тирозина вследствие пониженной активности фермента гомогентизиназы и накоплением в тканях организма гомотентизиновой кислоты; глазо-кожный альбинизм - обусловлен отсутствием синтеза фермента тирозиназы. Нарушения обмена углеводов галактоземия - отсутствие фермента галактозо-1-фосфат-уридилтрансферазы и накопление в крови галактозы; гликогеновая болезнь - нарушение синтеза и распада гликогена. Синдромы нарушения всасывания в пищеварительном тракте муковисцидоз; непереносимость лактозы Генные наследственные болезни: многообразие клинических проявлений: введение Симптоматика каждой генной болезни очень многообразна. Как правило, патологическим процессом затрагивается не одна система или орган, а несколько органов уже на первичных этапах формирования болезни. Это касается болезней, проявляющихся в нарушении процессов эмбрионального развития (врожденные пороки развития), наследственных болезней обмена веществ и комбинированных болезней. Например, при наследственных болезнях соединительной ткани нарушен синтез специфического для каждой болезни белка той или иной волокнистой структуры. Поскольку соединительная ткань есть во всех органах и тканях, то и многообразие клинической симптоматики при этих болезнях является следствием аномалии соединительной ткани. Так, при синдроме Марфана в патологический процесс вовлечены мышечно-скелетная система, глаза, сердечно-сосудистая система, наружные покровы, легкие, ЦНС, при синдроме Элерса-Данло - кожа, суставы, глаза, сердце, сосуды, грудная клетка, мозг, зубы. Наряду с понятными механизмами возникновения многообразия проявлений генных болезней имеются примеры необычайно широкого клинического проявления болезни с пока неизвестными механизмами. Нейрофиброматоз-1 проявляется пигментными пятнами, кожными, подкожными и плексиформными нейрофибромами, костными изменениями, опухолями нервных стволов и головного мозга, снижением способности к обучению. Эти многообразные проявления пока не удается связать в единый патогенетический комплекс, несмотря на то что уже известны структура гена и его первичный продукт. Не исключается, что в этом и других подобных случаях речь идет о первичной плейотропии, т. е. - множественных эффектах действия гена в разных органах. Генные болезни – это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. В зависимости от функциональной значимости первичных продуктов соответствующих генов генные болезни подразделяют на наследственные нарушения ферментных систем (энзимопатии), дефекты белков крови (гемоглобинопатии), дефекты структурных белков (коллагеновые болезни) и генные болезни с невыясненным первичным биохимическим дефектом. Энзимопатии. В основе энзимопатии лежат либо изменения активности фермента, либо снижение интенсивности его синтеза. У гетерозигот-носителей мутантного гена присутствие нормального аллеля обеспечивает сохранение около 50% активности фермента по сравнению с нормальным состоянием. Поэтому наследственные дефекты ферментов клинически проявляются у гомозигот, а у гетерозигот недостаточная активность фермента выявляется специальными исследованиями. В зависимости от характера нарушения обмена веществ в клетках среди энзимопатий различают следующие формы. 1. Наследственные дефекты обмена углеводов (галактоземия — нарушение метаболизма молочного сахара — лактозы; мукополисаха-ридозы — нарушение расщепления полисахаридов). 2. Наследственные дефекты обмена липидов и липопротеинов (сфинголипидозы — нарушение расщепления структурных липидов; нарушения обмена липидов плазмы крови, сопровождающиеся увеличением или снижением в крови холестерина, лецитина). 3. Наследственные дефекты обмена аминокислот (фенилкетонурия —нарушение обмена фенилаланина (см. разд. 4.1); тирозиноз— нарушение обмена тирозина; альбинизм — нарушение синтеза пигмента меланина из тирозина и др.). 4. Наследственные дефекты обмена витаминов (гомоцистинурия — развивается как результат генетического, дефекта кофермента витаминов В6 и B12, наследуется по аутосомно-рецессивному типу). 5. Наследственные дефекты обмена пуриновых и пиримидиновых азотистыхоснований (синдром Леша — Найяна, связанный с недостаточностью фермента, который катализирует превращение свободных пуриновых оснований в нуклеотиды, наследуется по Х-сцепленному рецессивному типу). 6. Наследственные дефекты биосинтеза гормонов (адреногенитальный синдром, связанный с мутациями генов, которые контролируют синтез андрогенов; тестикулярная феминизация, при которой не образуются рецепторы андрогенов). 7. Наследственные дефекты ферментов эритроцитов (некоторые гемолитические несфероцитарные анемии, характеризующиеся нормальной структурой гемоглобина, но нарушением ферментной системы, участвующей в анаэробном (бескислородном) расщеплении глюкозы. Наследуются как по аутосомно-рецессивному, так и по Х-сцепленному рецессивному типу). Гемоглобинопатии. Это группа наследственных заболеваний, вызываемых первичным дефектом пептидных цепей гемоглобина и связанным с этим нарушением его свойств и функций. К ним относят метгемоглобинемии, эритроцитозы, серповидно-клеточную анемию, талассемии (см. § 4.1). Коллагеновые болезни. В основе возникновения этих заболеваний лежат генетические дефекты биосинтеза и распада коллагена — важнейшего структурного компонента соединительной ткани. К этой группе относят болезнь Эллерса — Данлоса, характеризующуюся большим генетическим полиморфизмом и наследующуюся как по аутосомно-доминантному, так и по аутосомно-рецессивному типу, болезнь Марфана, наследующуюся по аутосомно-доминантному типу, и ряд других заболеваний. Наследственные болезни с невыясненным первичным биохимическим дефектом. К этой группе принадлежит подавляющее большинство моногенных наследственных болезней. Наиболее распространенными являются следующие. 1. Муковисцидозы — встречаются с частотой 1:2500 новорожденных; наследуются по аутосомно-рецессивному типу. В основе патогенеза заболевания —наследственное поражение экзокринных желез и железистых клеток организма, выделение ими густого, измененного по составу секрета и связанные с этим последствия. 2. Ахондроплазия — заболевание, в 80—95% случаев обусловленное вновь возникшей мутацией; наследуется по аутосомно-доминантному типу; встречается с частотой приблизительно 1:Это заболевание костной системы, при котором наблюдаются аномалии развития хрящевой ткани преимущественно в эпифизах трубчатых костей и костях основания черепа (рис. 6.23). 3. Мышечные дистрофии (миопатии) —заболевания, связанные с поражением поперечно-полосатых и гладких мышц. Различные формы характеризуются разным типом наследования. Например, прогрессирующая псевдогипертрофическая миопатия Дюшена наследуется по Х-сцепленному рецессивному типу и проявляется преимущественно у мальчиков в начале первого десятилетия жизни. Известна мышечная псевдогипертрофическая дистрофия, наследующаяся по аутосомно-рецессивному типу, которая начинает развиваться во второй половине первого десятилетия жизни и встречается с одинаковой частотой у обоих полов. Мышечная дистрофия плечевого и тазового пояса: наследуется по аутосомно-доминантному типу и т. д. Генетическое многообразие генных болезней. Изучение наследственных заболеваний у человека свидетельствует о том, что нередко сходное фенотипическое проявление болезни бывает обусловлено несколькими различными мутациями. Это явление впервые было описано в 30-х гг. С. Н. Давиденковым и названо генетической гетерогенностью наследственных заболеваний. Генетическая гетерогенность наследственных болезней может быть обусловлена мутациями разных генов, кодирующих ферменты одного метаболического пути, а также мутациями одного и того же гена, приводящими к появлению разных его аллелей. Среди рассмотренных выше наследственных болезней особенно высокой степенью генетического полиморфизма отличаются мукопо-лисахаридозы, генетическая разнородность которых объясняется множественными мутациями в 11—12 генах, связанных общей функцией расщепления полисахаридов. Большой генетической гетерогенностью характеризуется врожденная аутосомно-рецессивная форма глухоты, при которой различают не менее 35 генетически различных вариантов с фенотипически сходным проявлением. Большие перспективы в расшифровке наследственной гетерогенности генных болезней открываются в связи с применением молекулярно-генетических методов их прямого анализа с помощью ДНК-зондов. Есть моногенные и полигенные болезни. Моногенные болезни наследственного предрасположения – наследственные заболевания, проявляющиеся из-за мутации одного гена или проявляющиеся при действии определенного фактора среды (аутосомно-рецессивные или сцепленные с Х-хромосомой). Проявляются при воздействии факторов: - физических; - химических; - пищевых; - загрязнения среды. Парамиотомия – в сырую погоду происходят тонические спазмы мышц при холоде, под влиянием тепла – проходят. Болезнь связана с термочувствительным белком. Реакция проявляется в младенчестве и не изменяется на протяжении жизни человека. Пигментная ксеродерма - веснушчатая кожа особого типа. Проявляется в 4-6 лет. Дети не переносят УФ-свет возникают злокачественные опухоли, такие дети умирают от метастаз еще до 15 лет. Не переносят также и гамма-лучей. Синдром Блюма. Пигментная «бабочка» на лице, маленький рост, удлиненная голова. Евреи, поляки, беларусы, австрийцы. Погибают до 18 лет. Не переносят УФ-облучения, гамма-лучей. Альфа-1 антитрипсин при загрязнении воздуха, табачном дыме проявляется острой закупоркой бронхов или циррозом печени. У европеоидов люди, не переносящие молоко, составляют 10-20%, в Африке – 70-80%. Влияние лекарственных средств: сульфаниламидные препараты провоцируют заболевания крови. Есть полигенные болезни наследственного происхождения – такие болезни, которые возникают при действии многих факторов (мультифакториальные) и в результате взаимодействия многих генов. Установить диагноз в таком случае очень сложно, т. к. действует много факторов, и появляется новое качество при взаимодействии факторов. |