Тема 22. Ароматические альдегиды и кетоны. 2. Методы получения
 Скачать 206.89 Kb. Скачать 206.89 Kb.
|

|
2. Методы получения 1) Реакции окисления Аренкарбальдегиды получают окислением метиларенов и бензиловых спиртов. Окислители: CrO3 + (CH3CO)2O; CrO2Cl2 При окисление других алкиларенов образуются алкиларилкетоны. Окислители: О2 в присутствии катализаторов – солей кобальта и марганца. Окисление диарилметанов дает диарилкетоны. Окислители: KMnO4; CrO3. 2) Гидролиз дигалогеналкиларенов 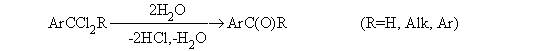 3) Ацилирование аренов 4) Формилирование аренов Для формилирования (введения группы СНО) необходим хлорангидрид муравьиной кислоты, который крайне нестабилен и разлагается на СО и НCl, поэтому используют другие формилирующие реагенты Реакция Гаттермана-Коха. Формилирование по Гаттерману-Коху осуществляется под действием оксида углерода (II) и хлористого водорода в присутствии катализатора Фриделя-Крафтса - хлорида алюминия, промотированного хлоридом меди (I). Роль однохлористой меди в этой реакции неясна: возможно она связывает СО в комплекс, что способствует образованию крайне нестабильного хлористого формила HCOCl из CO и HCl. Таким путем удается ввести альдегидную группу в алкилбензолы, арилгалогениды, полициклические углеводороды, причем формильная группа вводится селективно в пара-положение. Реакция не применима для формилирования фенолов, их эфиров и ариламинов. Реакция Гаттермана. В качестве формилирующего агента используется смесь безводного HCN и газообразного хлористого водорода в присутствии кислоты Льюиса. 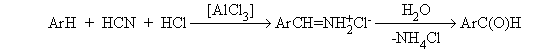 Электрофильным реагентом является, вероятно, комплекс катализатора с формимидхлоридом. 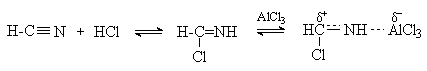 Образующийся в синтезе гидрохлорид альдимина ArCH=NH2+Cl гидролизуют до альдегида. Для того, чтобы избежать применения ядовитой синильной кислоты, Р.Адамс модифицировал реакцию, заменив синильную кислоту цианидом цинка (реакция Гаттермана-Адамса). Это позволило из цианида цинка и HCl получать непосредственно в реакционной смеси HCN и безводный хлористый цинк, играющий роль слабой кислоты Льюиса. Этот метод дает хорошие результаты при формилировании фенолов и простых эфиров фенолов. 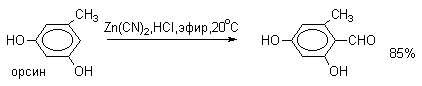 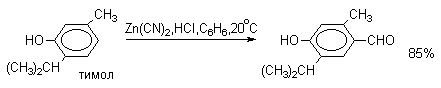 Реакции Гаттермана и Гаттермана-Адамса используют также для формилирования алкилбензолов и конденсированных ароматических углеводородов. Метод не применим для аренов, содержащих дезактивирующие заместители, и ароматических аминов. Реакция Вильсмейера-Хаака. В качестве формилирующего реагента используют диметилформамид (ДМФА) в присутствии POCl3 как кислоты Льюиса. Электрофильным агентом в реакции Вильсмейера-Хаака является иминиевая соль, которая образуется при взаимодействии ДМФА и хлорокиси фосфора (см. лек.29). 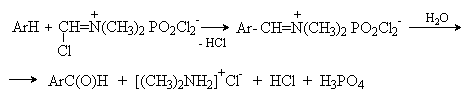 Реакция Вильсмейера-Хаака чрезвычайно проста в экспериментальном отношении и обеспечивает очень высокие выходы ароматических альдегидов, содержащих NR2-,OR- или OH-группы. Она оказывается практически ценной при формилировании конденсированных ароматических углеводородов - антрацена, азулена, пирена и др., а также разнообразных гетероциклических соединений ряда фурана, тиофена, пиррола, индола. Реакция Реймера-Тимана. Используется для формилирования фенолов. 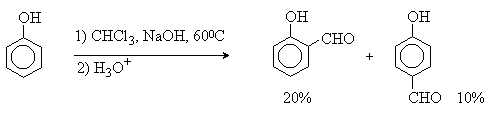 Подробнее см. лек.№29. 5) Восстановление хлорангидридов аренкарбоновых кислот (реакция Розенмунда) Реакция Губена — Гёша — метод синтеза арилкетонов ацилированием электроноизбыточных ароматических и гетероциклических соединений (фенолов и их эфиров, пирролов и т. п.) нитрилами в присутствии кислот Льюиса и хлороводорода: 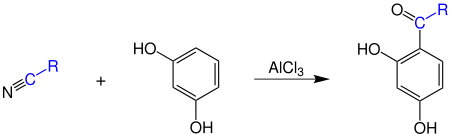 Является обобщением реакции Гаттермана-Коха, предложен в 1915 году Куртом Гёшем[1] и обобщён Йозефом Губеном[2]. Механизм В условиях реакции Губена — Гёша активирование нитрила идет за счет донорно-акцепторного связывания свободной электронной пары азота нитрила 1 с кислотой Люиса, в случае использования хлорида алюминия образующийся комплекс 2 может реагировать со второй молекулой AlCl3, образуя высокоэлектрофильный дикатионный интермедиат 3[3], после чего происходит ацилирование ароматического ядра, идущее по механизму SEAr с образованием кетимина 5, который далее гидролизуется до кетона 6:  3. Химические свойства Реакции ароматических карбонильных соединений подобны превращениям их алифатических аналогов. Однако необходимо отметить следующие особенности:
а) Бензоиновая конденсация При нагревании в присутствии цианидов аренкарбальдегиды образуют a -гидроксикетоны (бензоины). 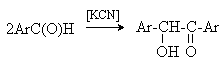 Механизм реакции: 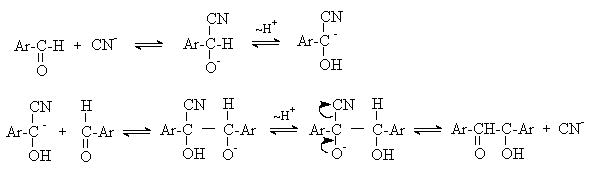 Ключевая стадия процесса – образование карбаниона за счет миграции протона от атома углерода к атому кислорода, которая становится возможной благодаря электроноакцепторному действию цианогруппы. б) Реакция Канницаро Аренкарбальдегиды, как альдегиды, не содержащие a -водородных атомов, вступают в реакцию Канницаро. 2ArC(O)H + NaOH ® ARCOONa + ARCH2OH Подробнее см. лек.№31-33. в) Реакция Перкина Аренкарбальдегиды конденсируются с ангидридами карбоновых кислот в присутствии оснований (ацетатов и карбонатов щелочных металлов, пиридина). При этом альдегид выполняет роль карбонильной, а ангидрид – метиленовой компоненты. Механизм реакции: 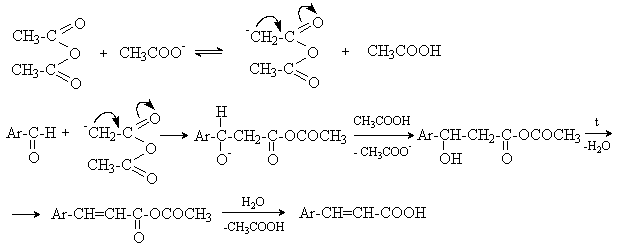 г) Автоокисление Аренкарбальдегиды очень легко окисляются кислородом воздуха на свету. ArCHO + 1/2O2 ® ArCOOH Процесс протекает по свободнорадикальному механизму через промежуточное образование стабильных ароильных радикалов Ar-C. =O. 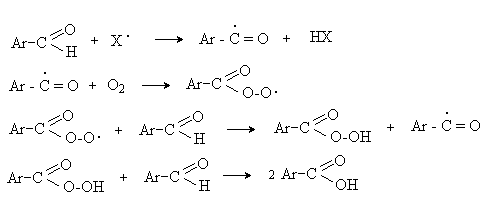 д) Хлорирование Аренкарбальдегиды легко хлорируются по свободнорадикальному механизму с образованием хлорангидридов. 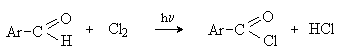 Механизм: 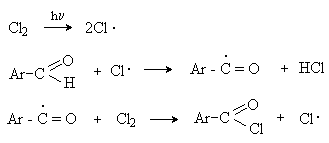 4. Важнейшие представители Бензальдегид– бесцветная жидкость с запахом горького миндаля. Получают из толуола прямым окислением или хлорированием до бензальхлорида с последующим гидролизом. Как альдегид, не содержащий a -водородных атомов, бензальдегид выступает в реакциях конденсации в качестве метиленовой компоненты. При конденсации его с ацетальдегидом образуется коричный альдегид: 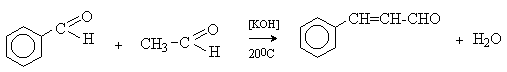 а при конденсации с с ацетофеноном – халкон: 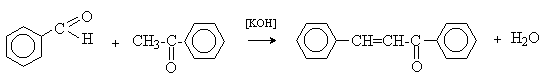 Коричный альдегид обладает свойствами a,b-непредельных карбонильных соединений. Используется в парфюмерии и пищевой промышленности. Бензофенон – бесцветное кристаллическое вещество. В промышленности его получают окислением дифенилметана. Для бензофенона характерно образование стабильного анион-радикала синего цвета при взаимодействии с металлическим натрием. 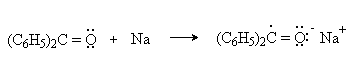 Такие анион-радикальные соли называют металлкетилами. Их относительная устойчивость связана со стабилизирующим влиянием на свободнорадикальный центр двух бензольных ядер и отрицательно заряженного атома кислорода. Дибензоил – желтое кристаллическое вещество. Образуется при окислении бензоина. 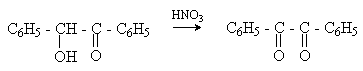 При нагревании со спиртовым раствором щелочи дибензоил претерпевает перегруппировку с образованием бензиловой кислоты (бензиловая перегруппировка). 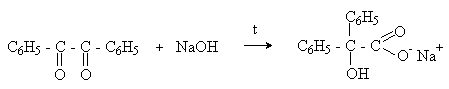 Механизм: 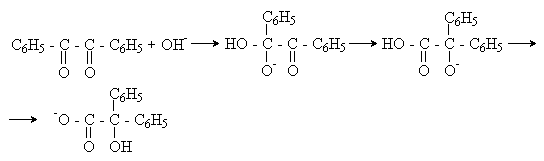 Бензоиновая конденсация (часто называемая реакцией конденсации, в силу исторических причин) — реакция между двумя ароматическими альдегидами, в частности, бензальдегидом. Реакция катализируется нуклеофилами, такими как анион цианида или N-гетероциклическими карбенами. Продукт реакции представляет собой ароматический ацилоин с бензоином в качестве исходного соединения. На первой стадии, цианид анион (образуемый от цианида натрия) вступает с альдегидами в реакцию нуклеофильного присоединения. Перегруппировка интермедиатов приводит к изменению полярности карбонильной группы, которая затем присоединяет вторую карбонильную группу посредством второго нуклеофильного присоединения. В результате переноса протона и элиминирования цианид иона образуется бензоин. Эта реакция обратима. 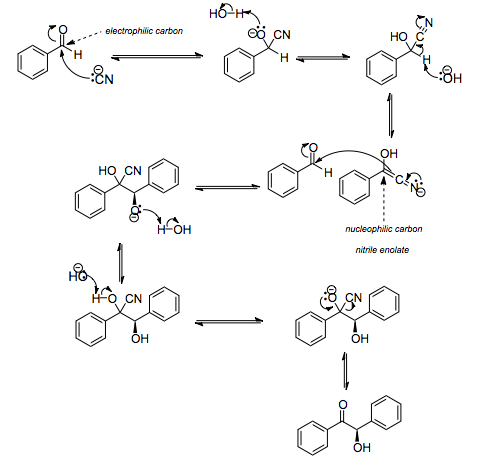 Цианид иона служит трем различным целям в этой реакции. Он действует как нуклеофил, облегчает отщепление протона, а также уходящей группы на заключительном этапе. В бензоиновая конденсация является скорее димеризацией, а не конденсацией потому, что малые молекулы, такие как вода, не выделяется в процессе этой реакции. По этой причине реакция называется также бензоиновое присоединение. В этой реакции два альдегида служат разным целям; один альдегид служит донором протона, а второй акцептором. Таким образом можно синтезировать смешанные бензоины, то есть продукты с различными группами на каждой стороне молекулы. 3.4. Ацилирование по Фриделю-Крафтсу Введение ацильной группы в ароматическое кольцо с помощью ацилирующего агента и кислоты Льюиса называют ацилированием по Фриделю-Крафтсу. Ацилирующими агентами обычно являются галогенангидриды и ангидриды кислот в присутствии галогенидов алюминия, трифторида бора или пентафторида сурьмы в качестве кислот Льюиса. Ацилгалогениды и ангидриды кислот образуют с кислотой Льюиса донорно-акцепторные комплексы состава 1:1 и 1:2. Спектральными методами было установлено, что хлорид алюминия, трифторид бора и пентафторид сурьмы координируются по карбонильному атому кислорода, так как он более основен чем соседний атом хлора. Электрофильным агентом в реакции ацилирования ароматических соединений является либо этот донорно-акцепторный комплекс, либо катион ацилия, образующийся при его диссоциации. 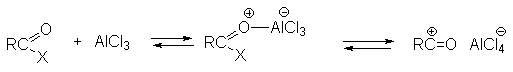 Координация по кислороду, а также образование катиона ацилия СН3СО+ доказано рентгеноструктурным анализом твердых комплексов ацетилхлорида с AlCl3 состава 1:1. Ацилгалогениды при взаимодействии с пятифтористой сурьмой в апротонной среде образуют ионно построенные соли RC+ =O SbF5X- , включающие катион ацилия. 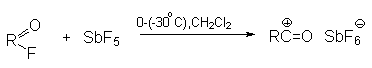 Можно полагать, что медленной стадией реакции является атака одного из трех электрофилов (RCO+ , RCOCl . AlCl3, RCOCl . Al2Cl6 ) на арен, приводящая к При ацилировании аренов ацилгалогенидами, катализируемом хлоридом или бромидом алюминия в полярных апротонных растворителях (нитробензоле, нитрометане и др.), ацилирующим агентом является катион ацилия, тогда как в малополярной среде (хлористом метилене, дихлорэтане или тетрахлорэтане) в реакции принимает участие донорно-акцепторный комплекс. Природа ацилгалогенида также оказывает влияние на образование и стабильность солей ацилия. Галогенангидриды ароматических карбоновых кислот легче превращаются в ацилиевые соли по сравнению с аналогами жирного ряда. Механизм реакции ацилирования аренов по Фриделю-Крафтсу под действием донорно-акцепторного комплекса описывается следующей схемой: 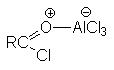 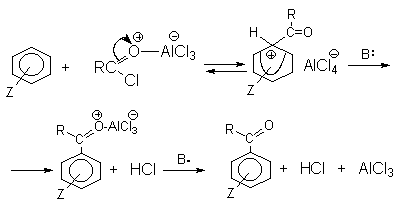 Ароматический кетон представляет собой более сильное основание Льюиса, чем ацилгалогенид и образует стабильный комплекс с AlCl3 или другой кислотой Льюиса. Поэтому для ацилирования ароматических соединений ацилгалогенидами требуется несколько больше эквимолярного количества катализатора, а при ацилировании ангидридами кислот два моля катализатора (т.к. они содержат два карбонильных атома кислорода). Кетон выделяют, разлагая его комплекс с AlCl3 водой или соляной кислотой. Ацилирование по Фриделю-Крафтсу полностью лишено тех недостатков, которые присущи реакции алкилирования. При ацилировании вводится только одна ацильная группа, поскольку ароматические кетоны не вступают в дальнейшую реакцию (так же, как и другие арены, содержащие сильные электроноакцепторные группы: NO2, CN, COOR). Еще одним преимуществом этой реакции по сравнению с алкилированием является отсутствие перегруппировок в ацилирующем агенте. Кроме того, для ацилирования не характерны реакции диспропорционирования продуктов реакции. Все эти особенности делают ацилирование по Фриделю-Крафтсу важнейшим методом синтеза жирноароматических и ароматических кетонов, которые получаются, как правило, с очень высоким выходами. Первоначально в качестве растворителя использовали сероуглерод, нитрометан, нитробензол или избыток жидкого ароматического углеводорода. В настоящее время предпочтение отдается тетрахлорэтану, 1,2-дихлорэтану и прежде всего легко летучему хлористому метилену, хорошо растворяющему хлорид и бромид алюминия. 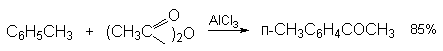 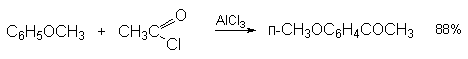 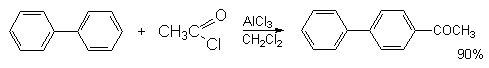 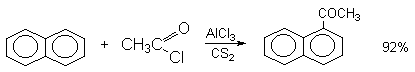 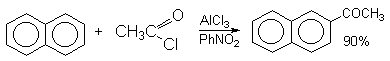 Возможно, такая селективность ацилирования нафталина связана с большим объемом комплекса CH3COCl . AlCl3 . PhNO2 по сравнению с комплексом CH3COCl . AlCl3 . CS2. Наиболее активными ацилирующими агентами являются смешанные ангидриды карбоновых кислот и трифторметансульфокислоты, обычно получаемые из ацилгалогенида и трифторметансульфокислоты непосредственно в реакционной смеси. Эти реагенты ацилируют бензол и другие ароматические углеводороды в отсутствие катализатора. 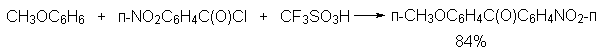 Ориентация входящей ацильной группы зависит от природы ацильной группы. Для хлоранангидридов и ангидридов алифатических кислот при реакции с аренами, содержащими заместители I-го рода, наблюдается очень высокая селективность замещения в пара-положение: 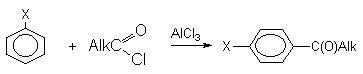 Соотношение орто/пара-изомеров в этом случае не превышает 0.03 и колеблется в интервале 0.01-0.03. Содержание мета-изомера также не превышает 0.5%. таким образом, ацилирование ароматических соединений хлорангидридами жирных кислот осуществляется чрезвычайно региоселективно в пара-положение. Доля орто-изомера резко возрастает при переходе к галогенангидридам ароматических карбоновых кислот. Для которых орто/пара-отношение изменяется в пределах 0.1 до 0.8. Эти данные находятся в хорошем соответствии с предположением о том, что для производных жирных кислот ацилирующим агентом является объемистый комплекс AlkCOCl . AlCl3, котрый атакует в ароматический субстрат в пространственно незатрудненное пара-положение. Меньшая селективность хлорангидридов ароматических кислот, возможно, объясняется тем, что в реакции принимает участие катион ацилия или его контактная ионная пара. Важное значение для синтеза бициклических и полициклических кетонов имеет внутримолекулярное ацилирование по Фриделю-Крафтсу. Имеется много вариантов этой реакции, некоторые наиболее типичные примеры приведены ниже. 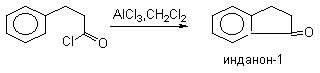 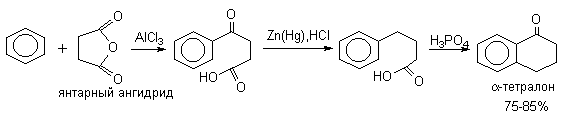 Халко́ны — соединения класса флавоноидов с незамкнутым пирановым кольцом. 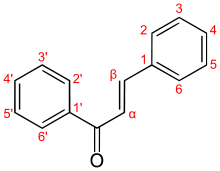 Структура халконов Соединения с насыщенной α-β-связью в пропановом фрагменте называются дигидрохалконами. Производные 1,2-дифенилпропана с незамкнутым пирановым кольцом носят название изохалканоиды, 1,1-дифенилпропана — неохалканоиды. Могут образовывать природные димеры — бихалканоиды. По наличию гидроксильных групп в кольце В могут быть незамещёнными (2',4'-дигидроксихалкон), моно-, дважды и трижды замещёнными. |