ЛАБОРАТОРНАЯ РАБОТА. ЛР6. Цель работы провести расчет переходного состояния и оценить энергию активации реакции. Привести энергетический профиль стереоизомерии. Порядок выполнения работы
 Скачать 0.75 Mb. Скачать 0.75 Mb.
|

|
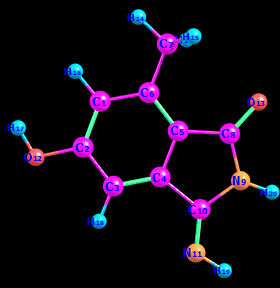
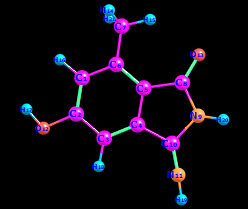
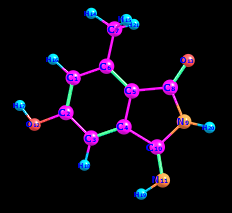
Цель работы: провести расчет переходного состояния и оценить энергию активации реакции. Привести энергетический профиль стереоизомерии. Порядок выполнения работы: Стереоизомеры, также пространственные изомеры — химические соединения, имеющие одинаковое строение, но отличающиеся пространственным расположением атомов. Дана реакция: Вариант №6.  В этой реакции меняется только диэдральный угол H-N-C-N. В результате такого поворота не появляется новое соединение, но появляется новый стереоизомер. Для определения вероятности такого поворота атома Н нужно определить энергию активации. Энергия активации (EA) – минимальное количество энергии, которое необходимо для протекания химической реакции. В ходе химического превращения система должна преодолеть потенциальный барьер. Поскольку пути реакции могут быть различны, то и высота этого барьера может изменяться. Геометрия молекулярной системы, соответствующая максимальной энергии системы на пути, имеющем самый низкий барьер, и получила название переходного состояния (ПС). 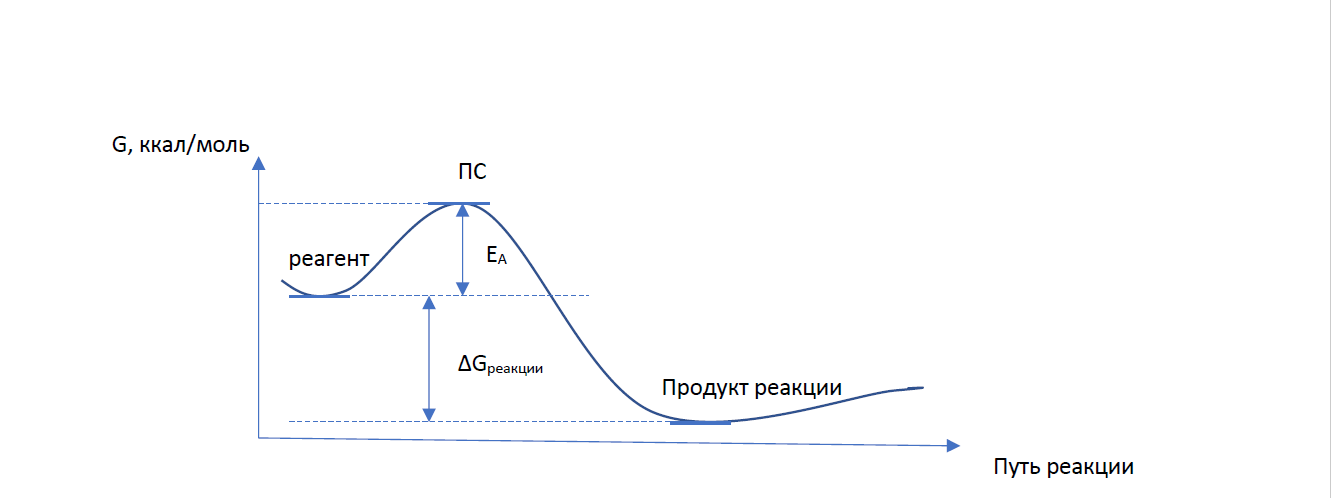 Рис. 1. Поверхность потенциальной энергии реакции. Переходное состояние или активированный комплекс–это состояние химической системы, промежуточное между исходными веществами и продуктам реакции. Рассмотрим модельную реакцию АВ+С↔[A...B...C] ↔А+ВС,протекающую так, что все атомы находятся на одной линии. С увеличением расстояния r(A-B) энергия системы возрастает до тех пор, пока не будет достигнута точка переходного состояния. В переходном состоянии [A...B...C] атом В уже частично связан с атомом С, но при этом он еще остается связанным с атомом А. Смещение от точки, соответствующей переходному состоянию, приводит к увеличению или уменьшению расстояний r(A-B) и r(B-С). При одновременном уменьшении длины одной связи и увеличении длины другой связи энергия понижается вплоть до образования продуктов или реагентов. Согласно постулату Хэммонда, если реакция эндотермическая , то структура переходного состояния должна быть близка к структуре продуктов реакции; если реакция экзотермическая – к структуре реагентов; если реакция теплонейтральная –структура переходного состояния лежит примерно посередине между структурами реагентов и продуктов. Часть I. Посчитаем структурные и энергетические характеристики реагентов, продуктов и переходного состояния реакции: 1.Создадим z-матрицу исходного соединения. Для расчета конечного стереоизомера в z-матрице исходного соединения изменим диэдральный угол атома Н. Каждое соединение рассчитывается отдельно! Расчеты будем проводить полуэмпирическим методом АМ1. 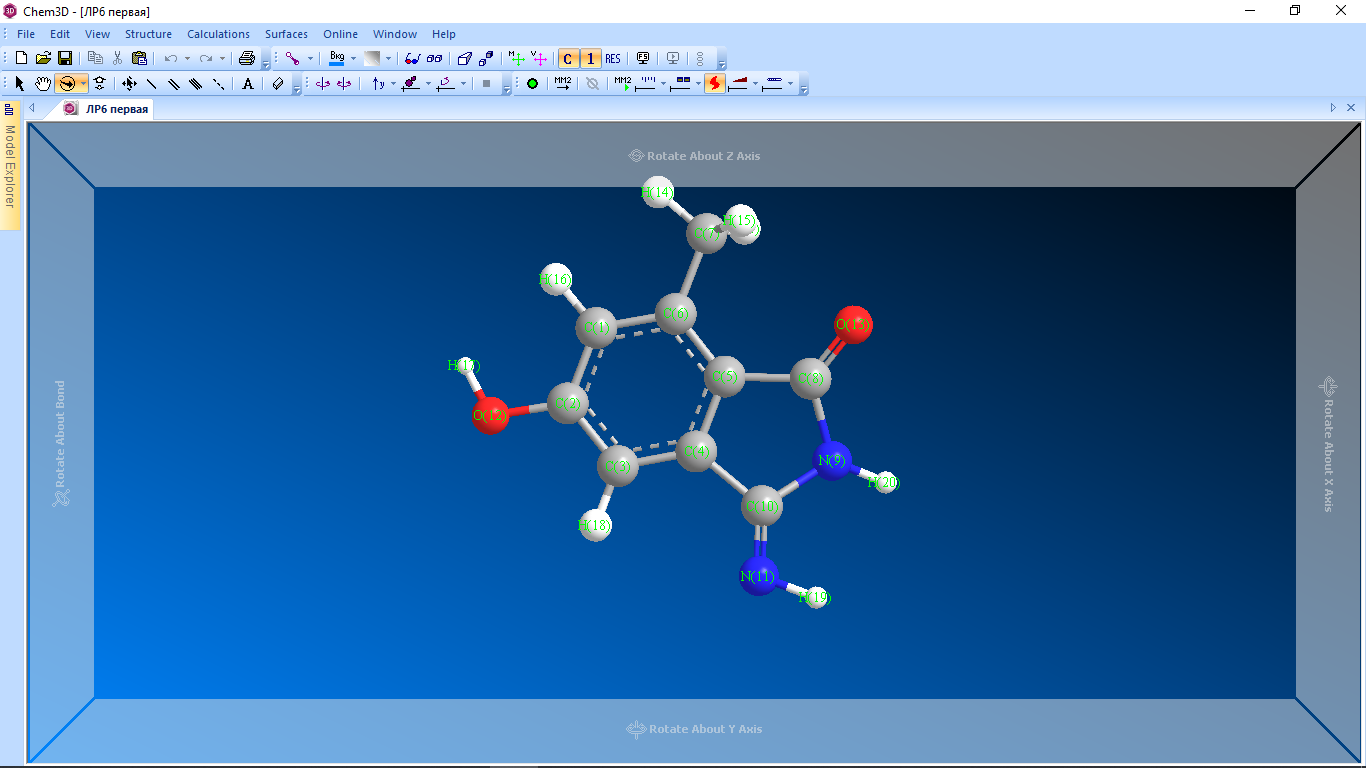 Рис 2. Трехмерная структура 1 молекулы в программе Chem3D, пронумеруем атомы. Запишем z-матрицу 1 молекулы: C C 1 1.405 C 2 1.397 1 120.6 C 3 1.384 2 118.1 1 -0.5 C 4 1.393 3 121.3 2 0.5 C 5 1.384 4 121.3 3 0.0 C 6 1.497 5 120.9 4 179.5 C 5 1.491 4 109.2 3 178.5 N 8 1.486 5 105.5 4 13.5 C 9 1.486 8 105.8 5 -21.4 N 10 1.260 9 127.3 8 -158.6 O 2 1.355 1 119.7 6 180.0 O 8 1.208 5 127.3 4 -166.5 H 7 1.113 6 109.5 5 180.0 H 7 1.113 6 109.4 5 -60.0 H 1 1.100 2 119.7 3 180.0 H 12 0.972 2 108.0 1 0.0 H 3 1.100 2 120.9 1 179.5 H 11 1.022 10 110.0 9 10.4 H 9 1.050 10 127.1 4 -158.6 H 7 1.113 6 109.5 5 60.0 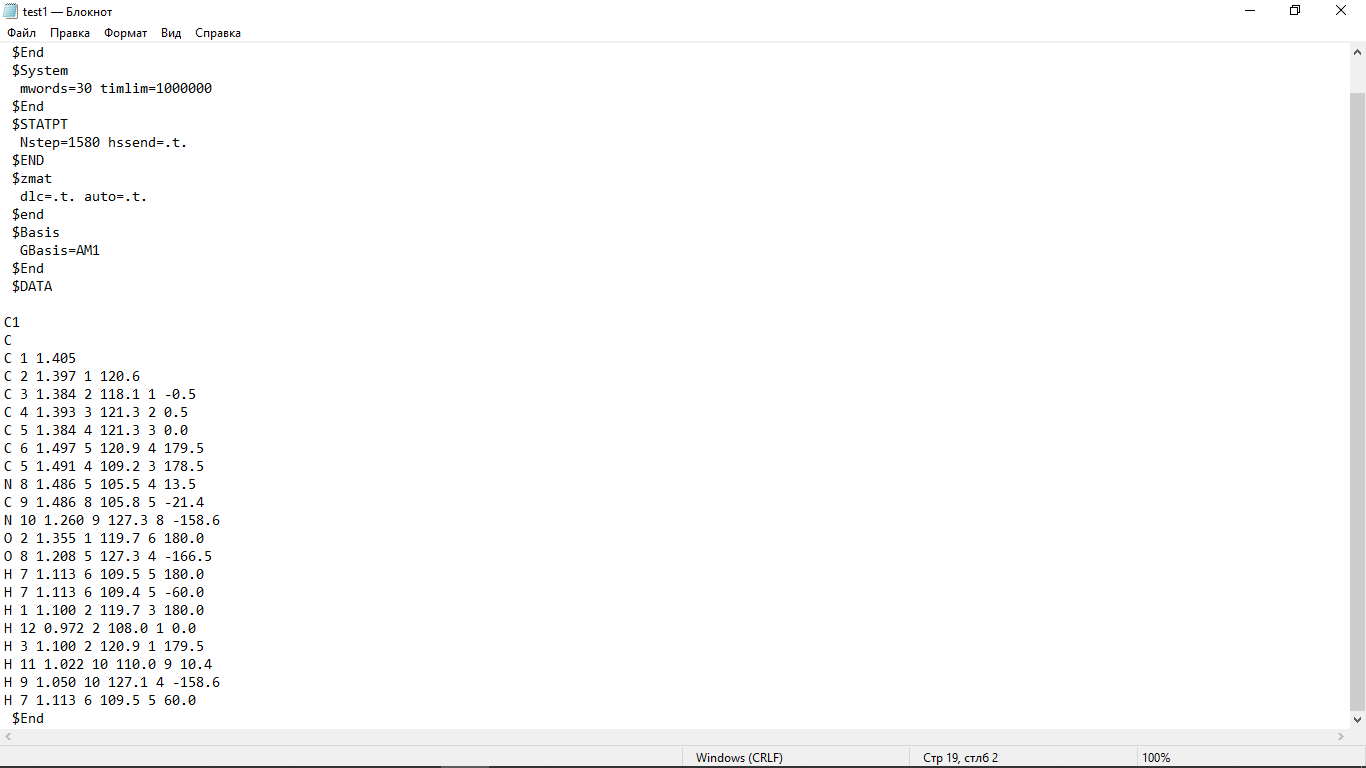 Рис. 3. Исходный код 1 молекулы. 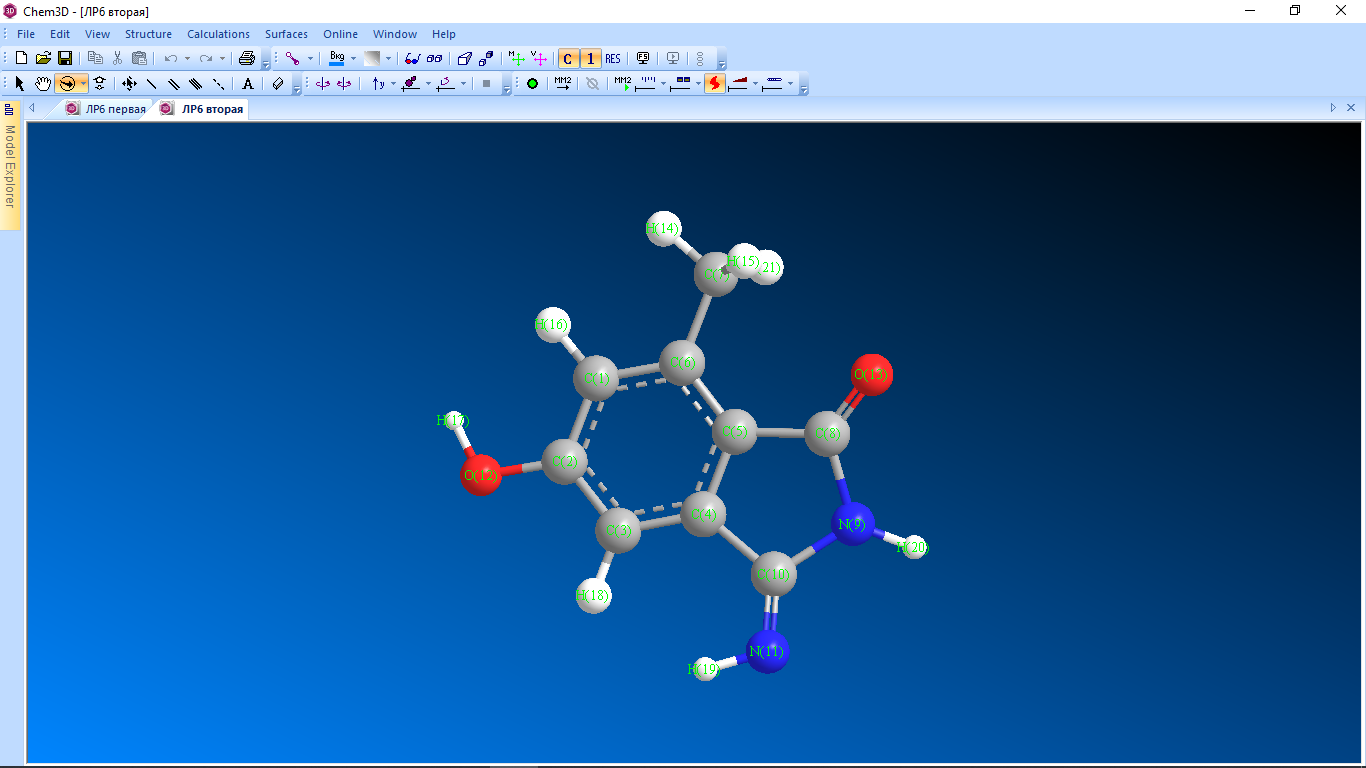 Рис 4. Трехмерная структура 2 молекулы в программе Chem3D, пронумеруем атомы. Запишем z-матрицу 2 молекулы: C C 1 1.405 C 2 1.397 1 120.6 C 3 1.384 2 118.1 1 -0.5 C 4 1.393 3 121.3 2 0.5 C 5 1.384 4 121.3 3 0.0 C 6 1.497 5 120.9 4 179.5 C 5 1.491 4 109.2 3 178.5 N 8 1.486 5 105.5 4 13.5 C 9 1.486 8 105.8 5 -21.4 N 10 1.260 9 127.3 8 -158.6 O 2 1.355 1 119.7 6 180.0 O 8 1.208 5 127.3 4 -166.5 H 7 1.113 6 109.5 5 180.0 H 7 1.113 6 109.4 5 -60.0 H 1 1.100 2 119.7 3 180.0 H 12 0.972 2 108.0 1 0.0 H 3 1.100 2 120.9 1 179.5 H 11 1.022 10 110.0 9 -180.0 H 9 1.050 10 127.1 4 -158.6 H 7 1.113 6 109.5 5 60.0 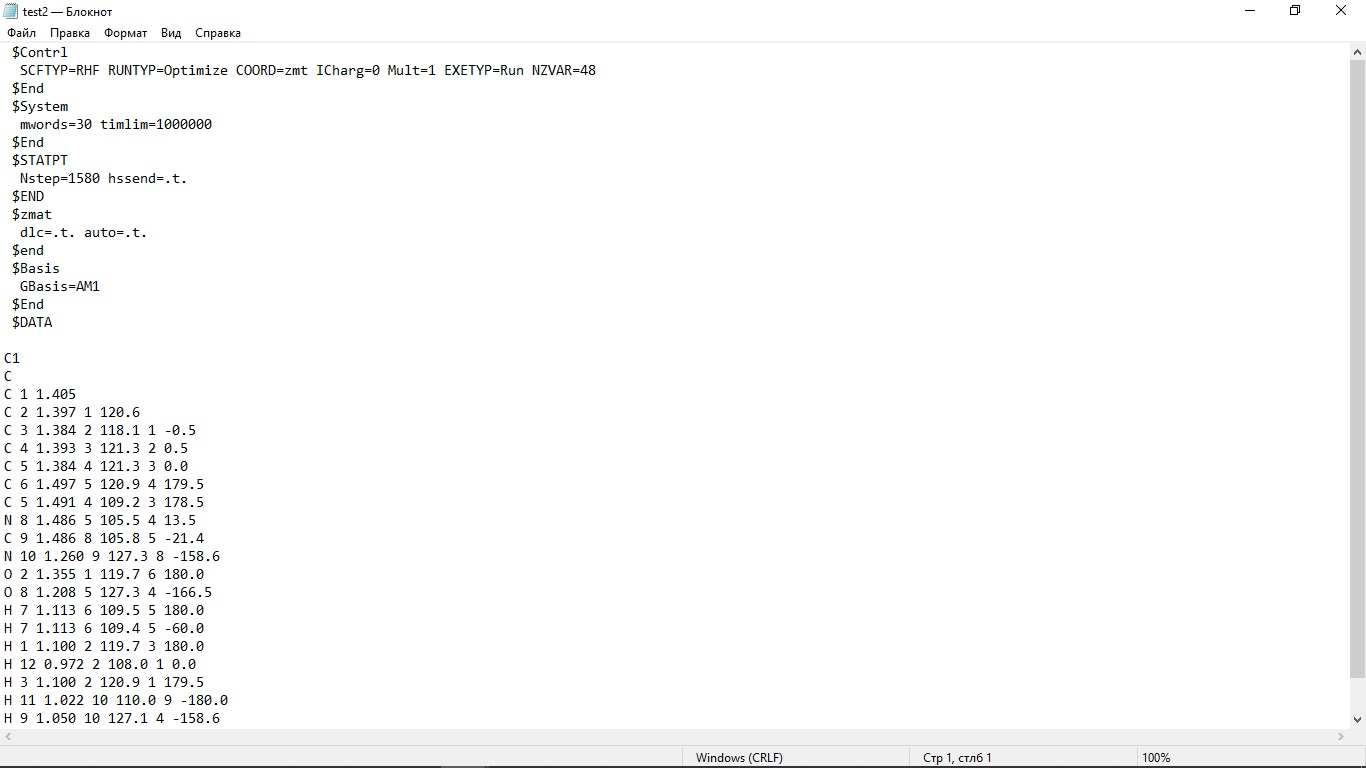 Рис. 5. Исходный код 2 молекулы. 2. Определим величину энергии Гиббса реакции как разницу между энергиями Гиббса продукта и реагента. Δ𝑟𝐺°=(Σ𝐸(𝑡𝑜𝑡𝑎𝑙)𝑃−Σ𝐸(𝑡𝑜𝑡𝑎𝑙)𝑅)∙𝑘+(Σ𝐺°𝑃−Σ𝐺°𝑅), где Σ𝐸(𝑡𝑜𝑡𝑎𝑙)𝑃 и Σ𝐸(𝑡𝑜𝑡𝑎𝑙)𝑅 – суммы полных энергий продуктов и реагентов соответственно; k – коэффициент пересчета Хартри в кДж/моль, равный 2625,500166; Σ𝐺°𝑃 и Σ𝐺°𝑅 - суммы температурных поправок по энергии Гиббса. Δ𝑟G°(298,15)= (-85,1193557540+85,122429802)*2625.500166 +(336,908-337,127) = =7,85191353 кДж/моль. В нашем случае она составила Δ𝑟G°=7,85191353 кДж/моль. 3. Поскольку значение энергии Гиббса реакции невелико, для определения переходного состояния процесса поворота атома водорода будем считать реакцию теплонейтральной, а значит, зададим переходное состояние как промежуточную структуру между реагентом и продуктом. Для этого достаточно задать атом Н в промежуточном положении между начальным и конечным положениями. 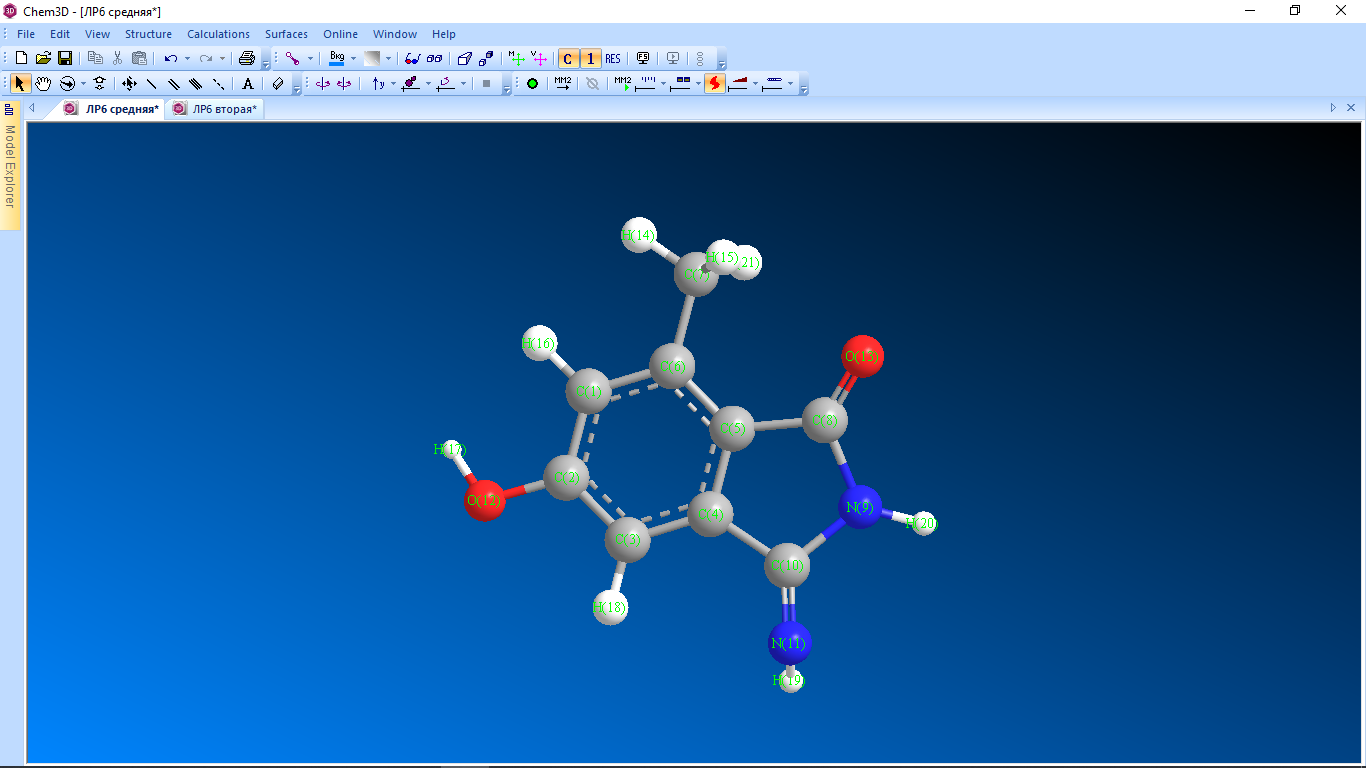 Рис 6. Промежуточная структура (переходное состояние) молекулы в программе Chem3D, пронумеруем атомы. Запишем z-матрицу переходной молекулы: C C 1 1.405 C 2 1.397 1 120.6 C 3 1.384 2 118.1 1 -0.5 C 4 1.393 3 121.3 2 0.5 C 5 1.384 4 121.3 3 0.0 C 6 1.497 5 120.9 4 179.5 C 5 1.491 4 109.2 3 178.5 N 8 1.486 5 105.5 4 13.5 C 9 1.486 8 105.8 5 -21.4 N 10 1.260 9 127.3 8 -158.6 O 2 1.355 1 119.7 6 180.0 O 8 1.208 5 127.3 4 -166.5 H 7 1.113 6 109.5 5 180.0 H 7 1.113 6 109.4 5 -60.0 H 1 1.100 2 119.7 3 180.0 H 12 0.972 2 108.0 1 0.0 H 3 1.100 2 120.9 1 179.5 H 11 1.022 10 110.0 9 -93.7 H 9 1.050 10 127.1 4 -158.6 H 7 1.113 6 109.5 5 60.0 Расчет проводится следующим образом: 1)Построенную структуру переходного состояния считаем аналогично расчетам реагента и продукта, только в группе параметров $Contrl меняется значение параметра RUNTYP на RUNTYP=sadpoint и добавляем в группу $STATPT параметр hess=calc . 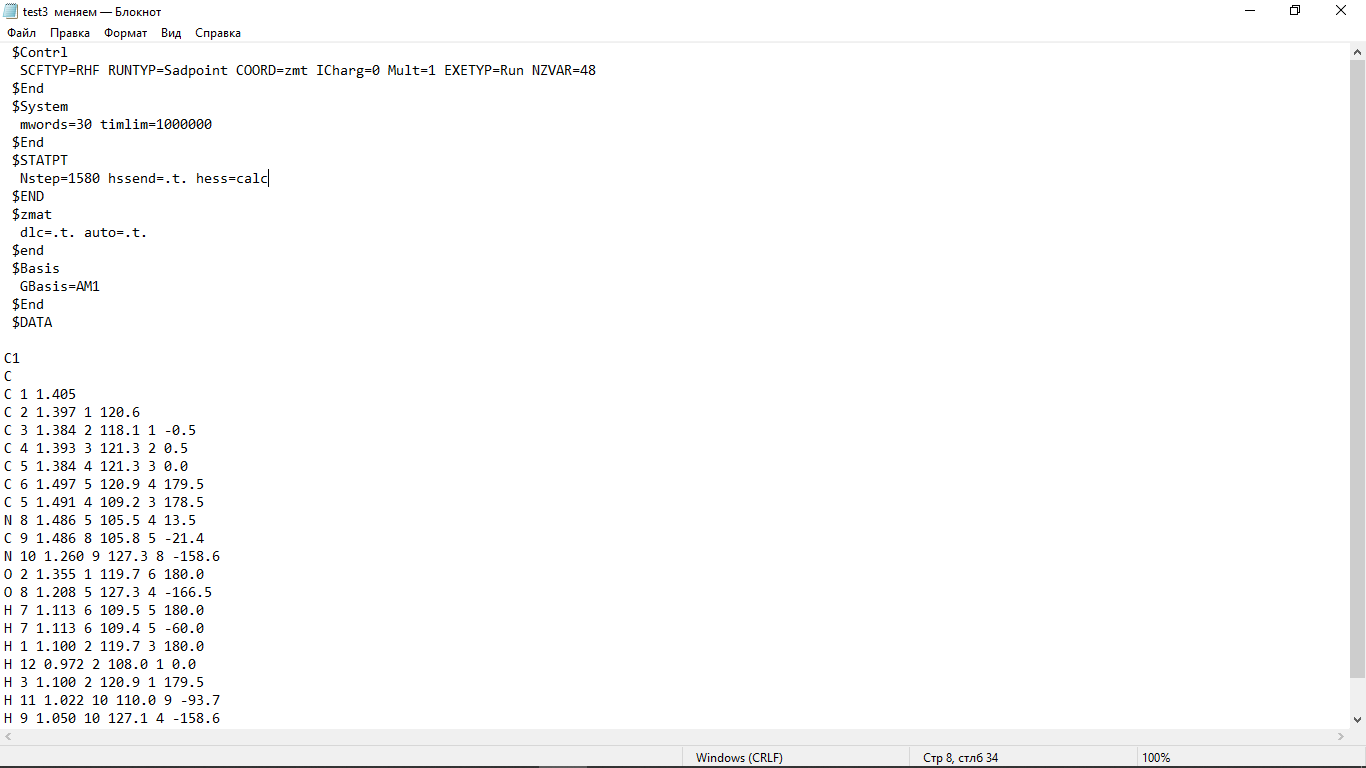 Рис. 7. Исходный код переходной молекулы. 2)Проверяем полученную структуру ПС на истинность: в столбце частот в программе Chemcraft должна быть одна отрицательная частота: 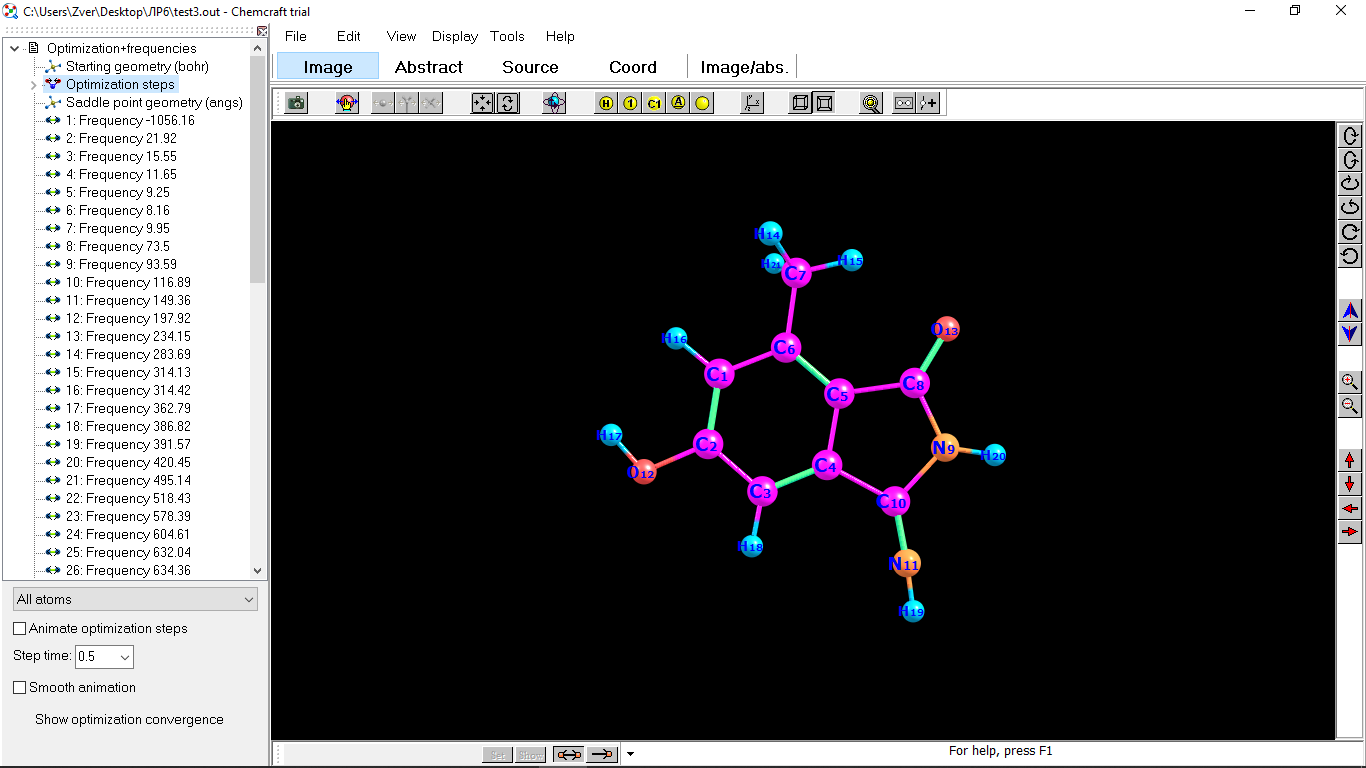 Рис. 8. Cтруктура переходной молекулы, в программе Chemcraft. 4. Результаты проведенных расчетов запишем в таблицу. Таблица №1. Результаты проведенных расчетов.
Посчитаем величину энергии Гиббса для всех соединений по формуле: G=E(total)*k+Gt Где E(total) – полная энергия; k – коэффициент перерасчета Хартри в кДж/моль, равный 2625,500166; Gt – температурная поправка по энергии Гиббса. G1=-85,122429802*2625,500166+337,127=-223151,827 кДж/моль Gперех.=-85,0887308918*2625,500166+326,868=-223073,609 кДж/моль G2=-85,1193557540*2625,500166+336,908=-223143,975 кДж/моль Энергия активации рассчитывается по формуле: ∆rG≠=G≠ - GR ∆rG≠=-223073,609+223151,827=78,218 кДж/моль Где G≠ - энергия Гиббса переходного состояния, GR - энергия Гиббса реагентов. Тогда: ∆rG≠= 78,218 кДж/моль 5. Построим энергетический профиль реакции: 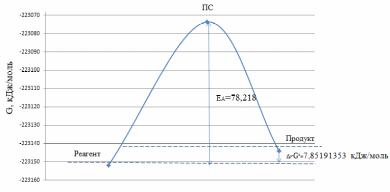 Рис. 9. Энергетический профиль реакции. 6. Вывод. Величина энергии активации: ∆rG≠= 78,218 кДж/моль Величина энергии Гиббса: Δ𝑟G°=7,85191353 кДж/моль Величина энергии активации на 70,3660865 кДж/моль больше, чем величина энергии Гиббса. Часть II. Докажем, что найденное переходное состояние соответствует исследуемому превращению. Для этого выполним расчет IRC (Intrinsic Reaction Coordinate) – расчет внутренней координаты реакции посредством градиентного спуска из ПС к исходным веществам и продуктам реакции вдоль пути реакции, что позволяет проследить, как изменяется при этом геометрия молекулы. Проверяют, приводит ли реакция, проходящая через данное переходное состояние, к нужным реагентам и продуктам. Исходный файл для реализации расчета IRC должен содержать координаты ПС и гессиан. Собственный вектор, соответствующий отрицательному собственному значению гессиана (отрицательная частота в списке частот), определяет координату реакции и направление спуска из ПС. Таких направлений должно быть два: в сторону исходных веществ и продуктов реакции. Для выполнения расчета за основу берётся файл расчета ПС. В группе параметров $Contrl меняем RUNTYP=sadpoint на RUNTYP=irc . В группе параметров $STATPT добавляем hess=read . Для выполнения расчета IRC в сторону продуктов реакции перед $data добавляем строку $irc npoint=10 saddle=.t. forwrd=.t. $end . Для выполнения расчета IRC в сторону реагентов перед $data добавляем строку $irc npoint=10 saddle=.t. forwrd=.f. $end . В качестве z-матрицы берем оптимизированную z-матрицу переходного состояния (ищем в файле расчета ПС по ключевому слову located). Далее после z-матрицы нужно добавить группу $HESS. Её нужно найти в PUNCH-файле (открываете PUNCH-файл в блокноте, ищете $HESS, копируете ВСЮ группу). |