перевод статьи. Cердечнососудистые заболевания (ссз) представляют серьезную угрозу для мирового общественного здравоохранения и продолжают вызывать растущую озабоченность
 Скачать 224.2 Kb. Скачать 224.2 Kb.
|

|
АННОТАЦИЯ Эффективный асимметричный синтез кальция аторвастатина был достигнут из коммерчески доступного диэтил-3-гидроксиглутарата с помощью нового подхода, который включает органокаталитическую энантиоселективную десимметризацию циклического ангидрида для установления стереогенности C(3) и сборки без цианида боковой цепи аминотипа C7 с помощью стратегии C5+C2 в качестве ключевых преобразований. 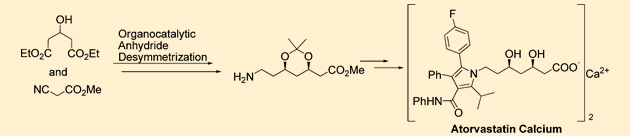 Cердечно-сосудистые заболевания (ССЗ) представляют серьезную угрозу для мирового общественного здравоохранения и продолжают вызывать растущую озабоченность. Сильная связь между уровнем холестерина липопротеинов низкой плотности (LDL-c), а также липопротеинов [LP(a)] и сердечно-сосудистым риском четко определена и подчеркивает их центральную роль в патогенезе атеросклеротической чумы.1 Лекарств, используемых для лечения гиперлипидемии, множество, но ни один класс лекарств не назначается так широко, как те, которые ингибируют 3-гидрокси-3-метил-глутарил кофермент А (ГМГ- КоА) редуктазы (статины).2 Причиной широкого применения статинов является их благоприятная эффективность, безопасные профили и долгосрочные клинические преимущества в снижении риска сердечно-сосудистых событий и смерти у пациентов с установленным ССЗ или без него. Кальций аторвастатин (Липитор, 1, рис. 1) был впервые представлен на рынке в 1997 году компанией Pfizer в качестве эффективного ингибитора ГМГ-КоА-редуктазы для лечения гиперхолестеринемии и атеросклероза и в настоящее время занимает первое место среди наиболее продаваемых лекарств в мире.3 На сегодняшний день совместные усилия по разработке синтетических продуктов привели к ряду стратегий, в которых используется хемоферментное разрешение4 или хиральное вспомогательное5 на соответствующей стадии, энантиоселективный катализ,6 или начиная с хирального пула.7 Однако, насколько нам известно, ни один из известных синтетических путей, по−видимому, не имеет коммерческого преимущества по сравнению с давно используемым подходом пиррола Пааля-Кнорра с использованием двух усовершенствованных строительных блоков дикетона 2 и боковой цепи 3 амино-типа C7 с рисунком син-1,3-диола, разработанным компанией Warner-Lambert в 1989 году (схема 1).7a−c Основной недостаток этого крупномасштабного синтеза заключается в манипулировании большим избытком высокотоксичного цианида при удлинении этого скелтона статина C7 через C3 + C1 + C2 + C1 маршрут (эквивалентом синтона C1 является NaCN или KCN). Кроме того, зависимость от одного синтетического процесса для поставки такого важного лекарственного средства является неразумной. В результате разработка альтернативных синтетических подходов к 3, в которых используется материал, не содержащий цианидов, для завершения асимметричного синтеза 1, является насущной необходимостью. Здесь мы сообщаем о новом, коротком, энантиоконтролируемом пути к 1, начинающемся с легкодоступного диэтил-3-гидроксиглутарата. 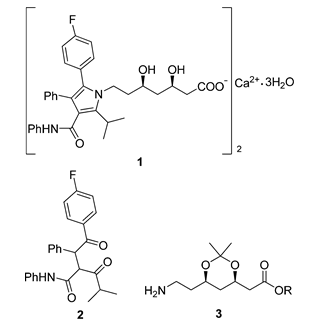 Рисунок 1. Структуры 1 и его важные промежуточные 2 и 3 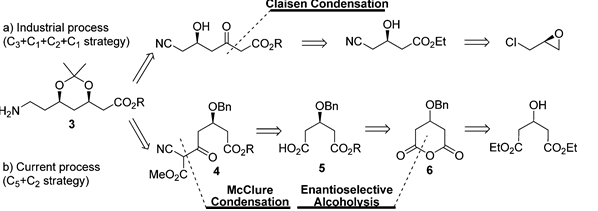 Схема 1. Описание производственного процесса и текущего процесса Наш подход к этой боковой цепи статина 3 был разработан с учетом того, что ее 3R-хиральность и боковая цепь аминотипа C7 могут быть собраны с помощью энантиоселективной десимметризации и стратегии C5 + C2 соответственно. Как указано в нашем плане синтеза (схема 1), ожидается, что энантиоселективный алкоголиз с использованием разработанных нами бифункциональных сульфонамидных катализаторов установит необходимую стереохимию 3R из циклического ангидрида 6, в то время как установка без цианидов боковой цепи 4 цианотипа C7 может быть достигнута путем конденсации хирального сложного эфира 5 (синтон C5) с метилцианоацетатом (синтон C2), который, в свою очередь, будет преобразован в 3 декарбоксилированием Крапчо, снятие защиты, сокращение Нарасаки, защита и последовательность сокращения. Учитывая важность энантиомерно чистого полуэфира 5 для успеха нашего запланированного асимметричного синтеза, мы искали эффективный метод получения желаемого 5. Имея в виду эту идею, мы обратили наше внимание на органокаталитическую энантиоселективную десимметризацию8 из 6 путем алкоголиза с использованием разработанных нами бифункциональных сульфонамидных катализаторов 9a−d.9, как показано на схеме 2, первоначально мы приготовили циклический ангидрид 6 из коммерчески доступного диэтил-3-гидроксиглутарата.Обработка диэтил 3-гидроксиглутарата бензилтрихлороацетимидатом в присутствии каталитического количества трифторметансульфоновой кислоты в циклогексане и метиленхлориде позволила получить соответствующий бензиловый эфир 7 с выходом 79%10, который подвергали LiOH-гидролизу и дегидратации ацетилхлоридом в метиленхлориде с получением 6 с общим выходом 91% (в течение двух стадий). 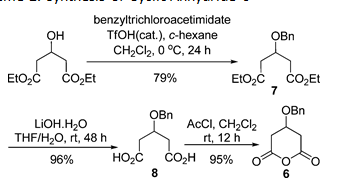 Схема 2. Синтез циклического ангидрида 6 Имея в руках 6, мы были готовы изучить желаемый каталитический энантиоселективный метанолиз в различных условиях реакции. Во−первых, разработанные нами бифункциональные сульфонамидные органокатализаторы 9a-d были проверены на предмет этой трансформации. Как видно из записей 1-4 в таблице 1, катализатор 9b обеспечивал требуемый полуэфир 5 с наивысшей энантиоселективностью (80% ee), когда реакцию 6 с избытком метанола проводили в метил-трет−бутиловом эфире (MTBE) в присутствии 5 мол.% органокатализаторов 9a-d при комнатной температуре. Влияние растворителя на энантиоселективность очень важно. Очень низкие энантиоселективности (42% и 36% ee) были обнаружены в метиленхлориде и ацетонитриле с использованием 5 мол.% 9b, а умеренные энантиоселективности (70%, 76% и 78% ee) были получены в толуоле, тетрагидрофуране и диэтиловом эфире соответственно (позиции 5-9). MTBE является лучшим выбором для этого метанолиза. Незначительное улучшение энантиоселективности (с 80% до 88% ee) наблюдалось при снижении концентрации субстрата с 0,1 до 0,0125 моль/л в присутствии 5 моль% 9b (записи 10-12). Незначительное увеличение энантиоселективности (90% ee) было достигнуто за счет увеличения загрузки катализатора с 5 до 10 мол. % (запись 13). Абсолютная конфигурация 5 была определена как требуемая R путем сравнения измеренного удельного поворота с представленными данными.11 Оставшимся ключевым этапом является удлинение боковой цепи 4 C7, содержащей цианофункциональную группу, из боковой цепи 5 C5. Обработка 5 (90% ee) метилцианоацетатом в присутствие диэтилпирокарбоната (DEPC) и избытка триэтиламина обеспечило диэфир 4 с выходом 86% в условиях реакции МакКлюра.12 Декарбоксилирование 4 во влажном ДМСО при 130°C плавно превращалось в кетоэфир 10 с выходом 79%. Расщепление бензилового эфира в 10 при обработке безводного треххлористого железа в безводном CH2Cl2 при 5°C в течение 48 ч приводило к образованию дебензильного продукта 11 с выходом 81% (схема 3). Таблица 1. Оптимизация условий Энантиоселективного метанолиза циклического ангидрида 60 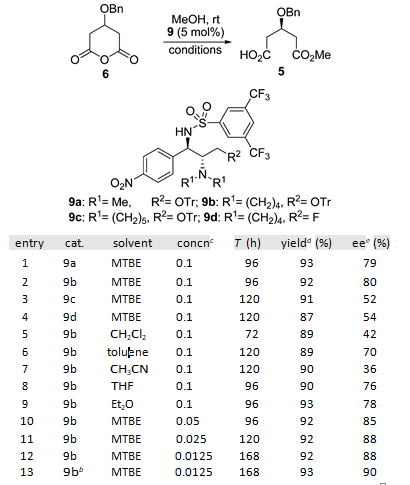 0 Если не указано иное, все реакции проводили с 6 (0,5 ммоль), 9 (0,025 ммоль) и MeOH (5,0 ммоль). B Загрузка катализатора составляла 10 мол. %. С концентрацией 6 (моль/л). Область применения изолированного продукта. Определяется методом ВЭЖХ. 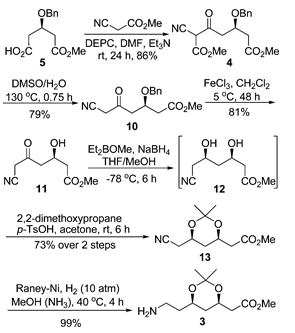 Схема 3. Синтез ключевого промежуточного продукта 3 Диастереоселективное восстановление 11 с использованием процедуры группы Нарасака 13 дало соответствующий диол 12, который затем был защищен 2,2-диметоксипропаном в присутствии п-толуолсульфоновой кислоты с получением 1,3-диоксана 13 с общим выходом 73% с превосходной диастереоселективностью [(4R,6R)-1,3-диоксан 13a/(4R,6S)-1,3-диоксан 13b = 95/5, определено методом ГХ−МС анализа). Стереохимия 13a и 13b, подтвержденная с помощью экспериментов NOESY, согласуется с механизм, предложенный Нарасакой и др. для восстановления ациклических β-гидроксикетонов с помощью хелата бора борогидридом натрия при стереоселективном получении 1,3-диолов (рис. 2). 13,16 Каталитическое гидрирование 13 смесью Raney-Ni и метанольного аммиака протекало плавно, обеспечивая желаемый амин 3 с почти количественным выходом. 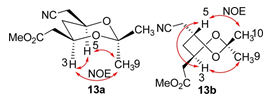 Рисунок 2. Корреляции NOESY 13a и 13b. Наконец, конденсацию 7c дикетона 2 и амина 3 по Паалю−Кнорру проводили в присутствии пиваловой кислоты при 90°C в течение 36 ч, чтобы получить пиррольное соединение 14 с выходом 74% (brsm 2) с 91% ee, которое было повышено до 99% ee после перекристаллизация с использованием EtOAc/PE (1:8) с восстановленным выходом 81%. Снятие защиты с 14 путем обработки соляной кислотой (1,0 М) в метаноле при комнатной температуре позволило получить диол 15 с выходом 94%. Гидролиз 15 гидроксидом натрия (1,0 М) в метаноле при 0°C и засоление 5%-ным водным ацетатом кальция при 0°C в последовательности 1 с общим выходом 91% в течение двух стадий (схема 4). Кроме того, мы определили кристаллическую форму 1 методом рентгеновской порошковой дифракции. Наблюдаемая кристаллическая форма соответствует форме I, сообщенной компанией Warner-Lambert. 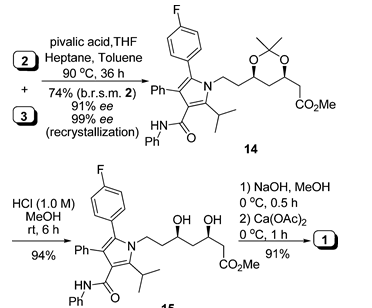 Схема 4. Синтез кальция аторвастатина (1) Заключение В заключение, стратегия десимметризации органокаталитического ангидрида и удлинения боковой цепи без цианидов, подробно описанная в этой статье, представляет собой эффективный и практичный процесс асимметричного синтеза кальция аторвастатина (1), начиная с легкодоступный и дешевый диэтил 3-гидроксиглутарат. Мы считаем, что наш подход принесет большую пользу для промышленного применения после дальнейшей оптимизации условий реакции на каждом этапе. |