Доклад Тромбофилии. Дневник по гематологии. Доклад по теме Тромбофилии
 Скачать 1.59 Mb. Скачать 1.59 Mb.
|

|
Министерство здравоохранения Российской Федерации Федеральное государственное бюджетное образовательное учреждение высшего образования Приволжский исследовательский медицинский университет Кафедра госпитальной терапии и общей врачебной практики имени В.Г. Вогралика Дневник практических занятий по циклу госпитальной болезни и эндокринология Цикл: Гематология Выполнила: Завидова Е.А., 648 гр. Проверил: Федотов В.Д., к.м.н., доцент кафедры госпитальной терапии и общей врачебной практики им. В.Г. Вогралика Нижний Новгород, 2022 г Доклад по теме «Тромбофилии» Тромбофилия - все наследственные (генетически обусловленные) и приобретенные (вторичные, симптоматические) нарушения гемостаза, которым свойственна предрасположенность к раннему появлению и рецидивированию тромбозов, тромбоэмболий, ишемий и инфарктов органов. Тромбофилия не является какой-либо болезнью, но представляет собой патологическое состояние, вызванное комбинацией постоянных и/или временных факторов риска, реализованных развитием тромбоза (тромбозов). Еще в 1884 году Рудольф Вирхов говорил о причинах тромбоза: Стаз крови в венах нижних конечностей Повышенная способность крови к тромбообразованию Повреждение стенки сосудов Нас больше интересует повышенная способность крови к тромбообразованию, поэтому может перейти к обсуждению факторов тромбогенного риска, которые могут быть врожденные и приобретенные, последние делятся на гематогенные и негематогенные: Врожденные Безусловно подтвержденные данные Дефицит антитромбина III Дефицит протеина С Дефицит протеина S Полиморфизм фактора V Лейден Полиморфизм протромбина (20210А) Гипергомоцистеинемия Подтвержденные данные Стойкое увеличение концентрации и/или активности: фибриногена, факторов II, VIII, IX или XI Дисфибриногенемия Гипоплазминогенемия и дисплазминогенемия Серповидно-клеточная анемия Приоретенные Негематогенные Операция, травма Активный рак Аутоиммунные заболевания Инфекция (пневмония, сепсис, инфекция мочевых путей, ВИЧ-инфекция) Дислипидемия, ожирение Нефротический синдром и вероятная хроническая почечная недостаточность Обезвоживание Беременность, послеродовой период Химиотерапия при лечении злокачественных новообразований Гормонотерапия АФС Гематогенные Гепарин-индуцированная тромбоцитопения Пароксизмальная ночная гемоглобинурия Тромботическая тромбоцитопеническая пурпура ДВС-синдром Для дифференциальной диагностики тромбофилий удобно использовать следующую схему 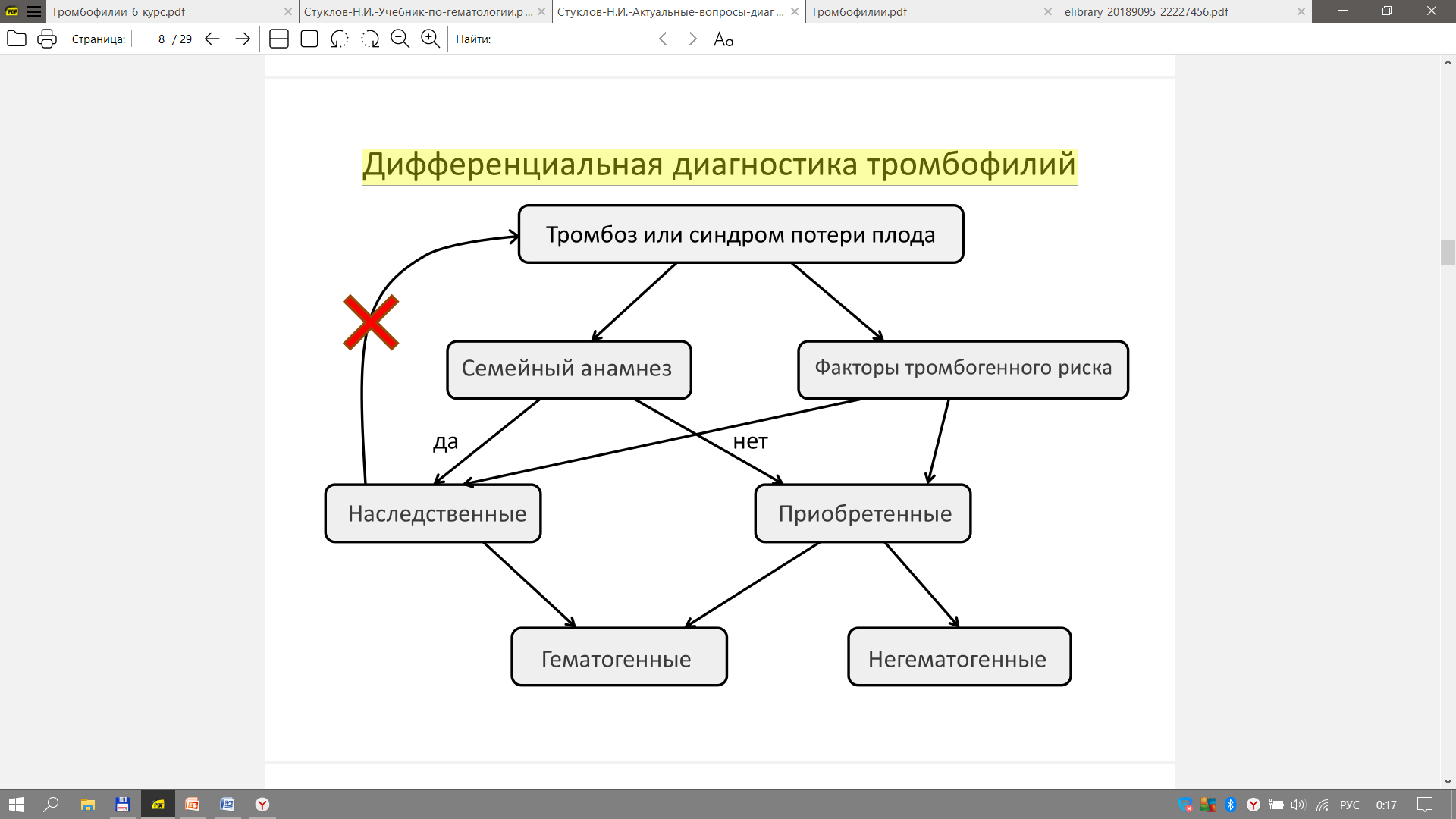 Классифицируют тромбофилии следующим образом: Гематогенные тромбофилии Аномалии сосудисто-тромбоцитарного гемостаза (тромбоцитозы, гиперагрегационный синдром, тромботическая тромбоцитопеническая пурпура) Дефицит, гиперпродукция или аномалии плазменных факторов свертывания крови (гиперфибриногенемии, повышенный уровень VII, VIII фактора, Лейденовская мутация V фактора, аномалия II фактора протромбина 20210А, наследственный дефицит XII фактора) Дефицит или аномалии физиологических антикоагулянтов (Антитромбин III, протеины С, S) Нарушение фибринолиза Метаболические (гипергомоцистеинемия) Негематогенные тромбофилии Гемореологические формы (сердечно-сосудистые заболевания, имобилизация, операции, травма…) Иммунные (воспалительное заболевание кишечника, ревматоидный артрит, системная красная волчанка, геморрагический микротромбоваскулит (болезнь Шенлейна–Геноха), гранулематоз Вегенера, АФС) Паранеопластические (рак, включая миелопролиферативные и миелодиспластические заболевания, синдром Труссо) Лекарственные (L-аспарагиназа, антиангиогенные препараты, гормоны, эритропоэтин, гепарины, эстрогены или прогестероны…) Ятрогенные формы Комбинированные формы Для диагностики тромбофилий используют рутинные лаборатоные исследования, которые дают четкую картину патологического состояния  Из всей популяции подвержено тромбофилиям не так уж и мало, около 15%, поэтому необходимо некоторым группам людей проводить диагностическое обследование. Вот следующие критерии для выборки обследуемых: Рекомендовано: Возраст моложе 45 лет Рецидивирующие тромбозы Не типичная локализация тромбоза Немотивированные тромбозы Родственники пациентов с установленной тромбофилией при подготовке к операции, перед назначением гормональной контрацепции, гормонотерапии На практике Все возрастные категории После первичного эпизода тромбоза Панель полиморфизмов генов свертываемости крови не обоснованно расширенa: FVII, FXIII, ITGA2, ITGВ2, PAI-I, FGB, МТГФР, МСР, МС . Большая группа пациенток с проблемами вынашивания беременности Беременные с «повышенными Д-Димерами» Невыполнение алгоритма диагностики АФС-синдрома Повышение активности прокоагулянтов Резистентность фактора Va к протеину С Описана впервые в 1993 г. шведом Бьерном Дельбеком – отсутствие ответа плазмы больного на добавление к ней активированного протеина С – АРС-резистентность (фактор V Leiden) 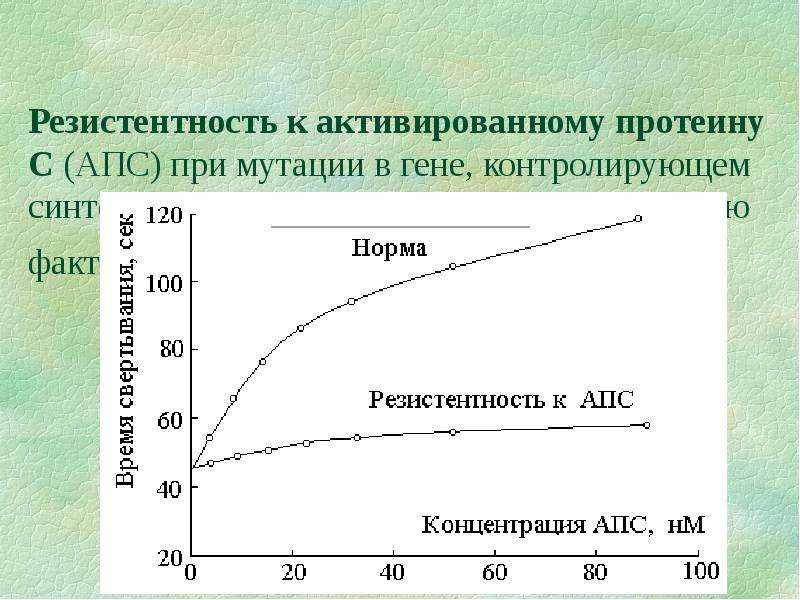 Эпидемиология: Встречается в основном среди представителей белой расы. Распространенность составляет 5–8%, достигая 15% в отдельных регионах (Греция, Швеция, Ливан). Частота данной мутации больше, чем дефицита антитромбина III, протеинов С и S вместе взятых. Этиология: Точечная мутация гена G1691A — замена гуанина (G) на аденин (А) в позиции 1691, в результате чего происходит замена аргинина (Arg506) на глутамин (Gln). Аутосомно-доминантный тип наследования. Клиническая картина: увеличение частоты возникновения венозных тромбозов, рецидивирующие флеботромбозы и тромбоэмболии Диагностика: генетическое исследование аномалии методом ПЦР; отсутствие увеличения АЧТВ при добавлении протеина Са по сравнению с контролем (отличия более чем в 2 раза). При гетерозиготном носительстве (А/G) вероятность клинических проявлений в 7 раз больше, чем в нормальной популяции, при гомозиготном (А/А) — в 80 раз. Однако, если этот фактор риска не взаимодействует с другими генетическими или приобретенными факторами риска, тромбоз может не возникнуть на протяжении всей жизни. Может сочетаться с дефицитом АТ III, протеина C и протеина S Профилактика: !!! Варфарин противопоказан низкие дозы ацетилсалициловой кислоты (75–375 мг/сут), клопидогрел (75 мг 1 раз в сутки), дипиридамол (25–75 мг 3–6 раз в сутки), пентоксифиллин (100–400 мг 2 раза в сутки), НМГ (при беременности): 0,2–0,6 мл для надропарина или 0,2–0,4 мл для эноксапарина п/к 1 раз в сутки Лечение: в острый период артериального или венозного тромбоза и ТЭЛА проводят трансфузии СЗП (800–1000 мл/сут) + гепарин под контролем АЧТВ; в подострый период — терапия сулодексидом и антиагрегантами (сулодексид внутрь по 250 мг 2 раза в сутки в течение 2–3 мес. + ацетилсалициловая кислота внутрь после еды 75–150 мг/сут длительно или клопидогрел внутрь 37,5 мг/сут (1/2 таблетки) длительно). Аномалия II фактора протромбина 20210А Эпидемиология: Встречается в основном среди представителей белой расы. Распространенность составляет 2–3%. Частота встречаемости у пациентов с тромбозами в 3 раза выше. Этиология: Ген протромбина располагается на хромосоме 11. Мутация гена протромбина G20210A характеризуется заменой нуклеотида гуанина нуклеотидом аденина в позиции 20210. Нуклеотидная последовательность измененного участка не участвует в кодировании аминокислотной последовательности гена протромбина, поэтому никаких химических изменений самого протромбина при наличии данной мутации не возникает. Аутосомно-доминантный тип наследования. Клиническая картина: увеличение частоты возникновения венозных тромбозов ранее развитие ИБС, ИМ, ТЭЛА Диагностика: повышение уровня протромбина (в 1,5–2 раза выше нормы) укорочение ПТВ Аномалия или гиперпродукция факторов VII и VIII Эпидемиология: Распространенность данного варианта в европейских популяциях составляет 10–20%. Этиология: Избыточное формирование комплекса тканевого фактора (Ф VIIа + Ф III) и повышенную активность Ф Х и Ф IX с последующим усилением выработки тромбина и формирования тромбов. Одно из проспективных исследований начала 1990-х гг. показало выраженную взаимосвязь между уровнем Ф VIIа и риском развития тромбоза. Ф VII (проконвертин) представляет собой неактивный витамин К-зависимый профермент, синтезируемый в печени и секретируемый ею в кровоток. Под действием тканевого фактора и последующим действием Ф Ха он переходит в активную форму, после чего формирует комплекс Ф VIIа/тканевой фактор, активирующий Ф Х и Ф IX. Ген, кодирующий проконвертин, имеет 5 полиморфных сайтов (участков), которые могут влиять на уровень циркулирующего фактора в крови. Проведенное в 1995 г. в Нидерландах контролируемое исследование показало, что отношение шансов по первичному эпизоду венозного тромбоза у лиц с уровнями Ф VIII свыше 150% составляет 4,8 по сравнению с лицами, у которых активность Ф VIII составляет менее 100%. Клиническая картина: Связана с повышенным риском смерти при развитии ИМ и тромбоэмболий. Диагностика: генетическое исследование аномалии методом ПЦР; увеличение количества определенного фактора свертывания укорочение АЧТВ (VIII), укорочение ПТВ (VII). Гиперфибриногенемия Гиперфибриногенемия – это состояние, связанное с повышением содержания фибриногена в крови. Эпидемиология: Распространенность в европейских популяциях составляет 5–10 %. Этиология: Чаще встречается при системных воспалительных и инфекционных заболеваниях, беременности — вторичная гиперфибриногенемия. Реже выявляют самостоятельную первичную форму, патогенез которой связан с наследственной мутацией гена фибриногена (полиморфизм G455A фибриногена). Замена гуанина на аденин в положении 455 приводит к повышенной производительности патологического А-аллеля, результатом которой становятся гиперфибриногенемия и высокий риск образования тромбов. Клиническая картина: повышенное АД, тромбоэмболические заболевания в анамнезе, инсульт. Диагностика: повышенный уровень фибриногена плазмы укорочение всех показателей коагулограммы Профилактика и лечение (факторы I, II, VII, VIII) Профилактика: длительная терапия варфарином 2,5–5 мг/сут под контролем МНО НМГ в профилактических дозах (привычное невынашивание беременности) Лечение по стандартной схеме: гепарин 5000 ЕД болюсно в/в, далее непрерывная инфузия 1000–1500 ЕД/ч (под контролем АЧТВ каждые 4–6 ч); перевод на НМГ: надропарин п/к 0,6–2 мл/сут (в зависимости от массы тела), или эноксапарин п/к 1–2 мг/кг/сут, или далтепарин по 100 МЕ/кг 2 раза в сутки, или фондапаринукс п/к 5–10 мг/сут в течение 7–10 дней; затем снижение дозы — поддерживающая терапия (1/2 дозы). Дефицит естественных антикоагулянтов Дефицит антитромбина III Самый сильный антикоагулянт c многофакторным действием (ингибирует факторы свертывания внешнего пути, тромбин). Впервые описан в 1965 г. О. Эгебергом в норвежской семье. Эпидемиология: Распространенность составляет 3-4%. Вторая по частоте среди наследственных тромбофилий. Этиология: Дефицит АТIII может быть наследственным и приобретенным (снижение синтеза при заболеваниях печени, повышение расхода при ДВС, оперативном вмешательстве, преэклампсии, кровотечении, лекарственной терапии гепарином, L-аспарагиназой, пероральными контрацептивами). Характер наследования — аутосомно-доминантный. Известно несколько типов дефицита АТ III: Тип I – сниженный биосинтез биологически нормальных молекул. Этот тип характеризуется снижением как антигенной, так и функциональной АТ-активности в крови носителя. У гетерозиготных носителей обе величины бывают снижены приблизительно на 50%. В основе типа I дефицита АТ могут лежать свыше 80 мутаций. Тип II характеризуется внутримолекулярными дефектами, т.е. нарушениями структуры молекул белка АТ. Проявляется он в том, что при нормальной иммунологической активности функциональная активность АТ резко снижена, что ведет к риску развития тромбоза. Наиболее распространен. Для типа III характерны нормальные уровни АТ (функциональный и антигенный) при нарушенном взаимодействии с гепарином, без которого АТ выполняет свою функцию крайне медленно. 70% больных со сниженной АТ-активностью имеют в анамнезе тромбоэмболические заболевания до достижения 50-летнего возраста. Для детского возраста клинические проявления дефекта не характерны. Клиническая картина: Наиболее частая локализация тромбозов – вены ног, таза, брыжеечные, артериальные тромбозы реже. Пик заболеваемости составляет 15–40 лет. Диагностика: снижение активности АТIII < 60 % в двух исследованиях Лечение: !!!Гепарин не эффективен. в острый период: концентрат АТIII в/в 1000–1500 ЕД/сут в первые 3–4 дня, далее 1 раз в 3 дня (содержание АТIII поддерживается выше 70 %); в подострый период: варфарин 2,5–5 мг/сут внутрь в одно и то же время суток под контролем МНО (2,0–3,5 еженедельно) длительно в качестве монотерапии или в сочетании с ацетилсалициловой кислотой 75–150 мг/сут внутрь после еды (или с клопидогрелом 37,5 мг/сут внутрь); МНО в таком случае не должно быть более 2,0. Дефицит протеина С и S Дефицит протеина С впервые описан в 1981 г. Дж. Гриффином в США, затем в Англии и Голландии, а дефицит протина S впервые описан в 1984 г. П. Компом и С. Эсмоном. Эпидемиология: Распространенность среди белого населения составляет 0,2–0,5%. Этиология дефицита C: Выделяют врожденные (наследственная тромбофилия) и приобретенные формы заболевания (дефицит витамина K, заболевания печени). Ген РС расположен на хромосоме 2 и тесно связан с геном Ф IX. Прямая функция этого белка – инактивация факторов свертывания Va и VIIIa. Оба эти фактора необходимы, в конечном счете, для адекватной выработки тромбина; их избыток ведет к стимуляции его выработки сверх нормы. Наследование аутосомно-доминантное. Наследственный дефицит протеина С может быть двух типов: I тип — уменьшение количества протеина С; II тип — снижение активности протеина С при его нормальном уровне. Этиология дефицита S: Протеин S является витамин K-зависимым фактором, синтезируется в печени, присутствует в двух формах — свободной и связанной. В норме 60–70 % протеина S связаны с С4-компонентом комплемента — регулятором классического пути системы комплемента. Уровень связывания протеина S с С4-компонентом комплемента определяет содержание свободного протеина S. Только свободная форма протеина S служит кофактором активированного протеина С. Наследственный дефицит протеина С может быть трех типов: Тип I – снижение концентрации свободного и общего протеина (чаще) Тип II – функциональный дефект молекулы протеина Тип III –концентрация общего нормальная, а свободного снижена (только свободная форма обладает антикоагулянтной активностью) Наследование аутосомно-доминантное. Гомозиготные варианты и сочетания с другими тромбофилиями обычно проявляются в период новорожденности с летальным исходом. Клиническая картина: Особенность этого дефекта состоит в том, что он может иметь клинические проявления уже в младенчестве. У гомозиготных носителей это может быть фульминантная (молниеносная) пурпура, у гетерозиготных – повышенный риск варфарин-индуцированного некроза кожи (в более позднем возрасте). При беременности дефицит РС может быть причиной тромбоза глубоких вен, преэклампсии, ограничения внутриматочного роста и рецидивирующих выкидышей. Первые тромботические эпизоды в 20-40 лет. Диагностика: снижение активности протеина С или S < 60 % в двух исследованиях Профилактика и лечение (дефицит протеина С и S) Профилактика: !!!Варфарин противопоказан сулодексид по 250 мг внутрь 2 раза в сутки в течение 2–3 мес.; при привычном невынашивании беременности — НМГ Лечение: в острый период: дротрекогин α (рекомбинантный активированный протеин С) в/в со скоростью 24 мкг/кг/ч в течение 96 ч; в подострый период: терапия сулодексидом c антиагрегантами (сулодексид по 250 мг внутрь 2 раза в сутки в течение 2–3 мес. + ацетилсалициловая кислота 75–150 мг/сут внутрь после еды длительно или клопидогрел 37,5 мг/сут внутрь длительно). Гипергомоцистеинемия Гипергомоцистеинемия – это дефект метилтетрагидрофолатредуктазы (МТГФР) или цистатион-b-синтетазы. Эпидемиология: Тяжелая гипергомоцистеинемия чаще развивается при гомозиготной форме заболевания. Наличие гомозиготы Т/Т выявляется у 10–16 % европейцев, а гетерозиготными носителями этого гена были 56 % обследованных лиц. Этиология: Наиболее изученной мутацией является вариант, в котором нуклеотид цитозина (C) в позиции 677 заменен тимидином (T), что приводит к замене аминокислотного остатка аланина на остаток валина (позиция 222) в сайте связывания фолата. Клиническая картина: гомоцистеинурия, эктопия хрусталика глаза, аномалии развития скелета, сосудистые заболеваниям в молодом возрасте до 30 лет (ИИ, ИМ), тромбоэмболии (ТЭЛА), когнитивные расстройства. Диагностика: содержание гомоцистеина в крови (15-30 – умеренная, 30-100 –промежуточная, более 100 мкмоль/л - тяжелая) определение методом ПЦР мутации с заменой нуклеотида цитозина на тимин в положении 677 (С677Т) в гене МТГФР; определение методом ПЦР мутаций других генов фолатного цикла. Профилактика: большие дозы фолиевой кислоты (5 мг/сут) или комплекс витаминов В6, В12, В9 внутрь в течение 2 мес. 3 раза в год под контролем содержания гомоцистеина в крови. Лечение: Острый период: гепарин + фолиевая кислота 5 мг/сутки Подострый период: НМГ, варфарин + фолиевая кислота 5 мг/сутки Клинический случай наследственной тромбофилии у больной язвенным колитом Больная И., 24 года. Считает себя больной с февраля 2015 г., когда появился жидкий стул до 6 раз в сутки с примесью слизи и крови. Обратилась в частный медицинский центр, где при колоноскопии в прямой, сигмовидной, нисходящей кишке отмечались отечность слизистой оболочки, отсутствие сосудистого рисунка, множественные дефекты слизистой оболочки до 2–3 мм, в восходящей и слепой кишке – единичные эрозии. Впервые поставлен диагноз: язвенный колит, тотальное поражение, впервые выявленный, легкой степени, умеренной активности. Назначено лечение месалазином 4 г в сутки, кишечными антисептиками. На фоне терапии положительной динамики не было. Обратилась в ФГБУ «ГНЦК им. А.Н. Рыжих» Минздрава России. К терапии добавлены микроклизмы с дексаметазоном 8 мг в сутки. При контрольной колонофиброскопии через 1 месяц наблюдалась положительная динамика в виде снижения активности воспалительных изменений до минимальной степени. Однако в июле 2015 г. стали беспокоить боли в левой нижней конечности, стул участился до 5 раз в сутки с примесью темно-красной крови. При ультразвуковой доплерографии сосудов нижних конечностей выявлен тромбоз глубоких вен левой нижней конечности, а при компьютерной томографии органов грудной клетки – признаки тромбоэмболии легочной артерии (крупных и мелких ветвей с обеих сторон). Госпитализация в ОРИТ, где проводилась инфузионная, антикоагулянтная, противогрибковая, антибактериальная, противовоспалительная терапия. Терапия микроклизмами с глюкокортикостероидами отменена. На фоне лечения сохранялся неоформленный стул 3–4 раза в сутки с примесью сгустков крови. 05.08.2015 выполнена имплантация кава-фильтра. При контрольной ультразвуковой доплерографии вен нижних конечностей от сентября 2015 г. отмечались посттромбофлебитические изменения, кава-фильтр проходим, наблюдались признаки реканализации тромба. Пациентка была обследована на тромбофилию, выявлены две гомозиготные тромбофилические мутации: 4G/5G в гене PAI-1 (ген ингибитора активатора плазминогена-1) – гомозиготная мутация; I/D полиморфизм в гене АСЕ (ген ангиотензинконвертирующего фермента). Установлен диагноз наследственной тромбофилии. Рекомендован прием комбинированного препарата диосмина и гесперидина (Детралекс) по 1 таблетке 2 раза в день 2 месяца, курсами 2 раза в год; эноксапарин натрия 0,4 мл или надропарин кальция 0,3 мл под контролем уровня тромбоцитов 1 раз в месяц, не менее 6 месяцев; прием системных и местных глюкокортикостероидов – только по жизненным показаниям. Для подбора противовоспалительной терапии пациентка в сентябре 2015 г. поступает в ГБУЗ МКНЦ им. А.С. Логинова ДЗМ с жалобами на боли в левой подвздошной области, кашицеобразный стул 3–4 раза в сутки с примесью сгустков крови, общую слабость. По данным колонофиброскопии диагностирован язвенный колит, левостороннее поражение: в сигмовидной и нисходящей кишке отмечена умеренная активность (рис. 1А), в прямой кишке – минимальная активность (рис. 1Б). При гистологическом исследовании слизистой оболочки толстой кишки обнаружены характерные признаки язвенного колита умеренной активности (рис. 2). 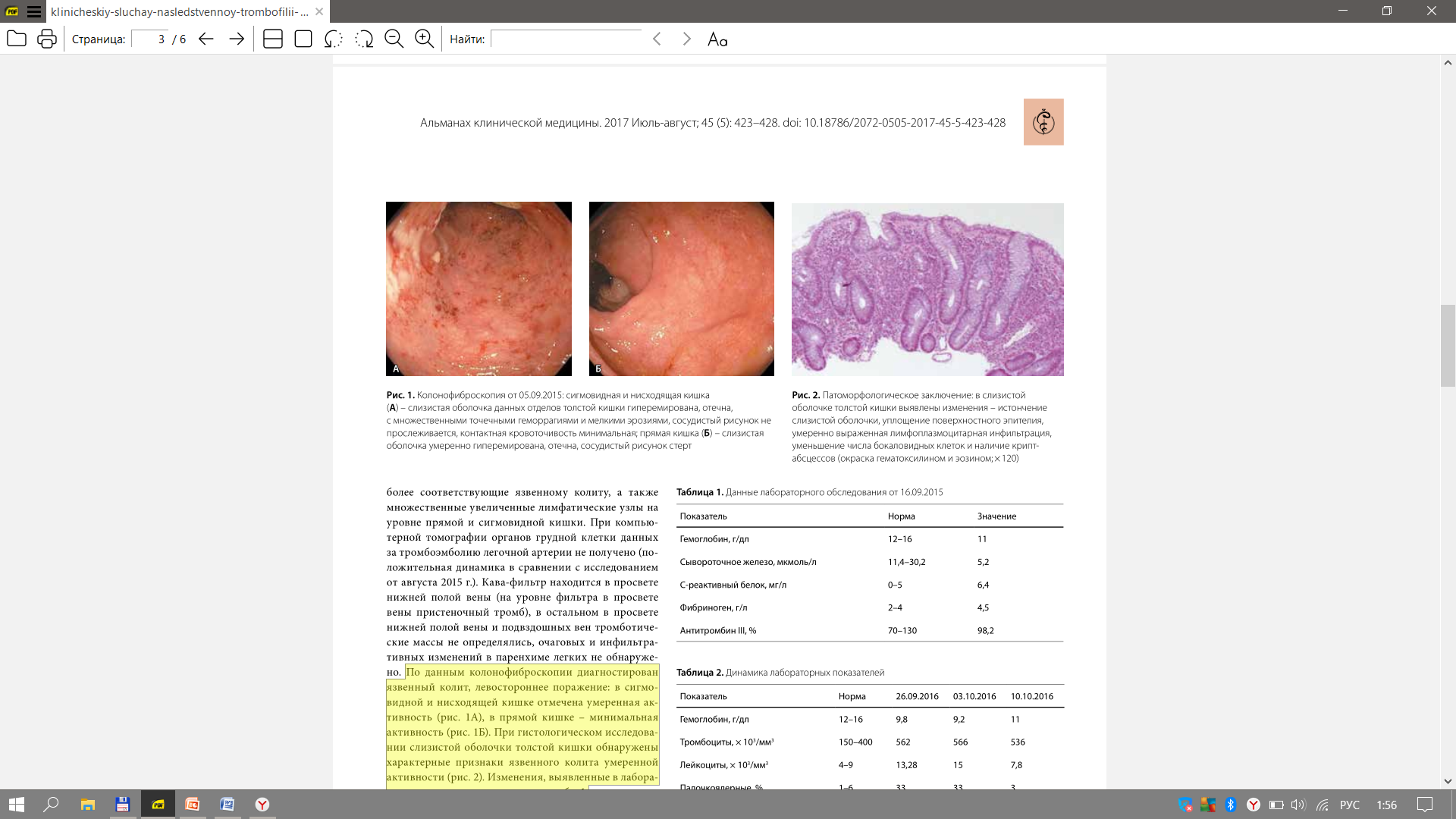 Изменения, выявленные в лабораторных показателях, отражены в табл. 1. Тромбоэластограмма показала наличие признаков умеренной гиперкоагуляции: снижение активированного времени свертывания (R) – 4,2 минуты (норма 5–10), повышение активности фибриногена – 73,4° (норма 53–72), повышение агрегации тромбоцитов (МА) – 71,7 мм (норма 50–70), повышение коагуляционного индекса (Cl) – 3,5 (норма -3–3), повышение прочности сгустка (G) – 12,7 °/с (норма 4,6–10,9). Рекомендован постоянный прием пероральных антикоагулянтов (дабигатран) 75 мг в сутки (с учетом массы тела пациентки 45 кг). В стационаре проведено лечение: микроклизмы с месалазином 2 г в сутки, месалазин 4 г в сутки, препараты железа внутримышечно, антибактериальная терапия – с положительным эффектом в виде уменьшения болей в животе, урежения стула до 2 раз в сутки, отсутствия примеси крови. В качестве постоянной противовоспалительной терапии пациентке рекомендован прием месалазина 4 г в сутки длительно, микроклизмы с месалазином 2 г в сутки 1 месяц, затем 2 раза в неделю длительно, прием антикоагулянтов под наблюдением гематолога по месту жительства. Далее больная по собственному решению антикоагулянтную терапию не принимала, у сосудистого хирурга наблюдалась нерегулярно, дозу месалазина снизила самостоятельно до 3 г в сутки, местную терапию месалазином проводила нерегулярно. В сентябре 2016 г. пациентка повторно поступает в ГБУЗ МКНЦ им. А.С. Логинова ДЗМ с жалобами на учащенный стул до 5 раз в сутки, больше в вечерние часы, кал полуоформленный, периодически с примесью крови, спастические боли по ходу ободочной кишки перед дефекацией. Результаты проведенного лабораторного обследования (26.09.2016) даны в табл. 2. По тромбоэластограмме определены признаки выраженной гиперкоагуляции: повышение активности фибриногена – 77,7° (норма 53-72), агрегации тромбоцитов (МА) – 77,4 мм (норма 50-70), коагуляционного индекса (Cl) – 4 (норма -3-3), прочности сгустка (G) – 17,1 °/с (норма 4,6-10,9). 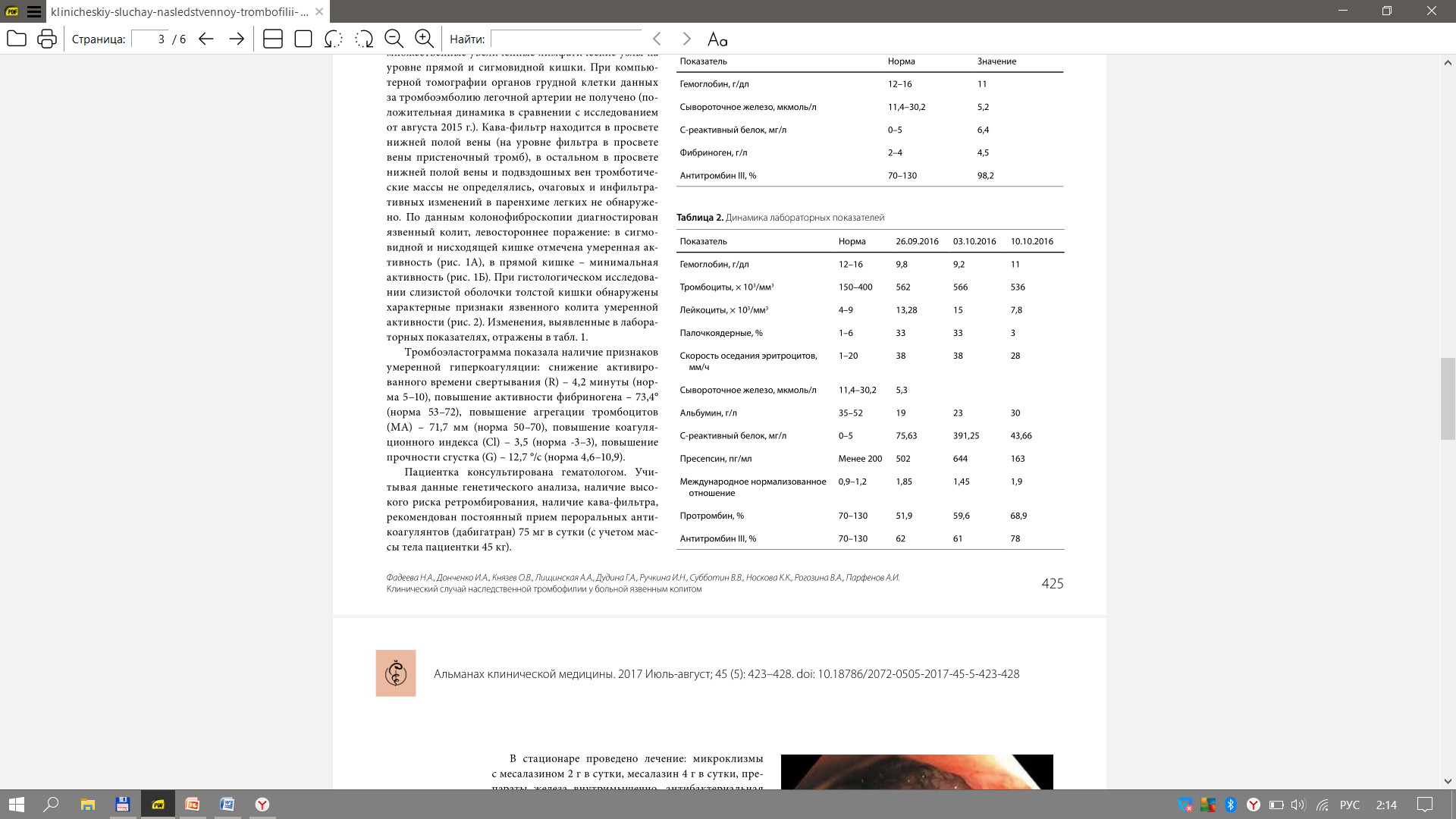 Заключение по данным колонофиброскопии (без подготовки): язвенный колит, тотальное поражение, высокая степень активности (рис. 3). Проведено лечение: метронидазол 1 г в сутки внутривенно, цефтриаксон 2 г в сутки внутривенно, месалазин 4 г в сутки, препараты железа внутримышечно, трансфузия свежезамороженной плазмы, альбумин 20% 100 мл внутривенно 1 раз в сутки. На фоне терапии через 7 дней сохранялся жидкий стул до 10 раз в сутки с примесью алой крови в каждой порции, повышение температуры до 38,5 °С. В лабораторных показателях (03.10.2016) также отмечалась отрицательная динамика. 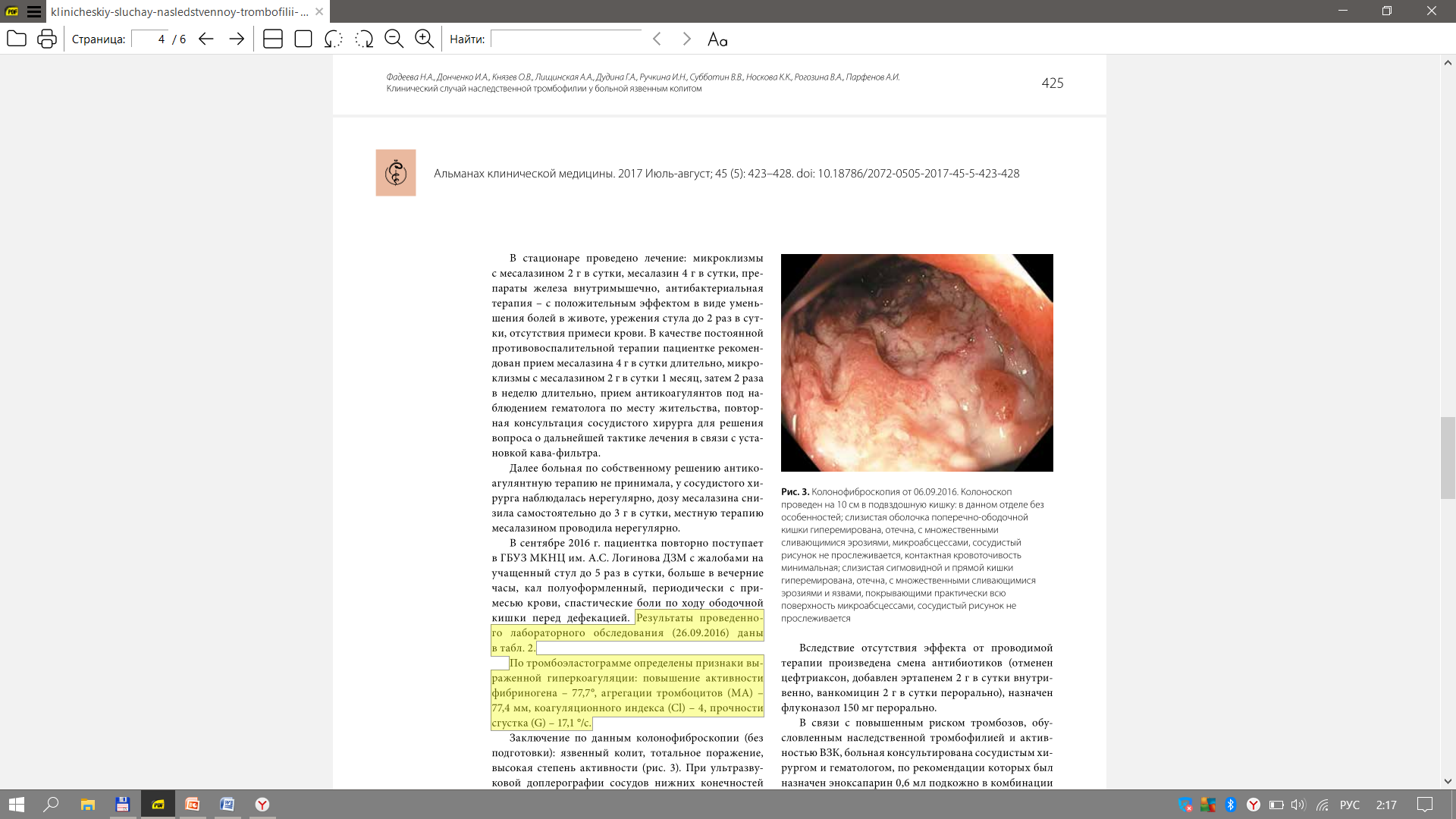 Поскольку у пациентки были два отягощающих друг друга заболевания (язвенный колит высокой активности и наследственная тромбофилия) и отсутствовал эффект от проводимой терапии, был проведен мультидисциплинарный консилиум (в составе гастроэнтерологов, колопроктологов, реаниматологов, гематологов), на котором принято решение о необходимости выполнения колопроктэктомии с формированием постоянной илеостомы. Однако от хирургического лечения пациентка отказалась. По этой причине, а также с учетом высокой активности и протяженности воспаления принято решение о назначении системной гормональной терапии преднизолоном 60 мг в сутки внутривенно капельно в комбинации с эноксапарином 0,4 мл подкожно 2 раза в сутки и индукционным курсом адалимумаба по стандартной схеме 160 мг – 80 мг – 40 мг. На фоне терапии через 10 дней у больной уредился стул до 4–5 раз в сутки, примесь крови в стуле исчезла. По лабораторным данным (10.10.2016) отмечалась положительная динамика. В тромбоэластограмме на фоне терапии надропарином зарегистрирована нормокоагуляция. Установлен клинический диагноз: язвенный колит, тотальное поражение, хроническое непрерывное тяжелое течение, высокой степени активности. Белково-энергетическая недостаточность 2–3-й степени. Дефицит массы тела (индекс массы тела 16). Хроническая железодефицитная анемия средней степени тяжести. Наследственная тромбофилия. Тромбоз вен левой нижней конечности, тромбоэмболия ветвей легочной артерии от 28.07.15, имплантация кава-фильтра от 05.08.15. Посттромботические изменения подколенной вены слева. Далее пациентка была выписана на амбулаторное лечение с рекомендациями продолжить антицитокиновую терапию адалимумабом в дозе 40 мг подкожно каждые 14 дней; прием месалазина 4 г в сутки – длительно, прием глюкокортикостероидов по схеме с постепенным снижением дозы каждые 7 дней до полной отмены, микроклизмы с месалазином 2 г в сутки, надропарин подкожно 0,6 мл 2 раза в сутки под наблюдением гематолога и сосудистого хирурга и контролем коагулограммы и тромбоэластограммы. Список литературы и Интернет-ресурсов Тромбозы и тромбофилии: классификация, диагностика, лечение, профилактика / С. А. Васильев, В. Л. Виноградов, А. Н. Смирнов [и др.] // РМЖ. Медицинское обозрение. – 2013. – Т. 21. – № 17. – С. 896-901. – EDN QZUTGN. Клинический случай наследственной тромбофилии у больной язвенным колитом / Н. А. Фадеева, И. А. Донченко, О. В. Князев [и др.] // Альманах клинической медицины. – 2017. – Т. 45. – № 5. – С. 423-428. – DOI 10.18786/2072-0505-2017-45-5-423-428. – EDN ZUCHCX. Учебник по гематологии / Н.И. Стуклов, Г.И. Козинец, Н.Г. Тюрина. — М.: Практическая медицина, 2018. — 336 с. - ISBN 978-5-98811-492-5. Лекция «ТРОМБОФИЛИЯ: диагностика и лечение. Место и роль лабораторной диагностики»; ГБУЗ МО МОНИКИ им. М.Ф.Владимирского Центральная клиническая лаборатория. Кафедра клинической лабораторной диагностики. Лекция «Тромбофилии и антифолсфолипидный синдром»; ПСПбГМУ им. Павлова. Кафедра госпитальной терапии. Лекция «Актуальные вопросы диагностики и лечения тромбофилий»; Стуклов Н.И.; ФГАОУ ВО Российский университет дружбы народов; кафедра госпитальной терапии. |