Обмен липидов. Функции липидов жестко связаны с их строением
 Скачать 1.24 Mb. Скачать 1.24 Mb.
|

Холестерол жизненно необходим клеткам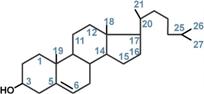 Холестерол относится к группе соединений, имеющих в своей основе циклопентан-пергидрофенантреновое кольцо, и является ненасыщенным спиртом. ИсточникиСинтез холестерола в организме составляет примерно 0,5-0,8 г/сут, при этом половина образуется в печени, около 15% в кишечнике, оставшаяся часть в любых клетках, не утративших ядро. Таким образом, все клетки организма способны синтезировать холестерол. Из пищевых продуктов наиболее богаты холестеролом (в пересчете на 100 г продукта) сметана (0,002 г), сливочное масло (0,03 г), яйца (0,18 г), говяжья печень (0,44 г). В целом за сутки с обычным рационом поступает около 0,4 г. Выведение из организмаВыведение холестерола из организма происходит в основном через кишечник:
Функции холестерола1. Структурная – входит в состав мембран, обуславливая их вязкость и жесткость. 2. Связывание и транспорт полиненасыщенных жирных кислот между органами и тканями в составе липопротеинов низкой и высокой плотности. Примерно 1/4 часть всего холестерола в организме этерифицирована олеиновой кислотой и полиненасыщенными жирными кислотами. В плазме крови соотношение эфиров холестерола к свободному холестеролу составляет 2:1. 3. Является предшественником желчных кислот, стероидных гормонов (кортизола, альдостерона, половых гормонов) и витамина D. Болезни накопления липидов называются липидозыГенетические заболевания, при которых происходит неполное расщепление полимерных веществ и их накопление, получили название лизосомные болезни накопления, так как они обусловлены дефектами специфических лизосомальных гидролаз. В случае накопления липидов такие болезни называются липидозы. При липидозах нарушается нормальный катаболизм липидов до соответствующих мономеров. ЛипидозыБолезнь Вольмана – редкое аутосомно-рецессивное заболевание из-за дефекта кислой эстеразы лизосом, что обусловливает накопление эфиров холестерола в лизосомах печени, селезенки, надпочечников, костного мозга и тонкого кишечника. Проявляется в первые недели жизни рвотой, диареей и стеатореей, гепатоспленомегалией и двусторонним кальцинозом надпочечников. Больные умирают в возрасте до 6 мес. Болезнь Шюллера-Кристиана аутосомно-рецессивное заболевание характеризуется отложением в плоских костях, твердой мозговой оболочке и коже холестерола и его эфиров. Характерными являются деструктивные изменения в костях, остеопороз, мозжечковые расстройства. Заболевание обычно начинается в возрасте до 10 лет, реже позднее. Мужчины болеют в 2 раза чаще, чем женщины. Течение заболевания прогрессирующее. Дефектный фермент неизвестен. Болезнь Гоше – отложение цереброзидов в макрофагальных клетках селезенки, печени, лимфатических узлов и костного мозга. Возникает в связи с аутосомно-рецессивным отсутствием гликоцереброзидазы (β-глюкозидазы). Основными симптомами заболевания являются спленомегалия, увеличение печени и селезенки, а также изменения в костях, проявляющиеся в виде остеопороза.  Дефектный фермент при болезни ГошеПри болезни Нимана-Пика наблюдается отложение сфингомиелина в клетках различных органов из-за дефицита сфингомиелиназы. Болезнь наследуется аутосомно-рецессивно, проявляется резким увеличением печени и селезенки, замедлением психического развития ребенка, появлением слепоты и глухоты. Чаще всего дети погибают в возрасте до 2 лет.  Дефектный фермент при болезни Нимана-ПикаБолезнь Тея-Сакса (амавротическая семейная идиотия) является результатом дефекта N-ацетилгексозаминидазы, при котором происходит отложение ганглиозидов в клетках головного мозга, что сопровождается атрофией зрительных нервов, слепотой, слабоумием и смертью в младенческом возрасте. 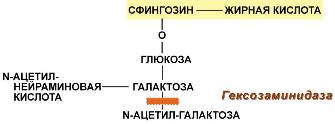 Дефектный фермент при болезни Тея-Сакса |