Лекции по белкам биохимия 2005. Кафедра биохимии
 Скачать 1.2 Mb. Скачать 1.2 Mb.
|

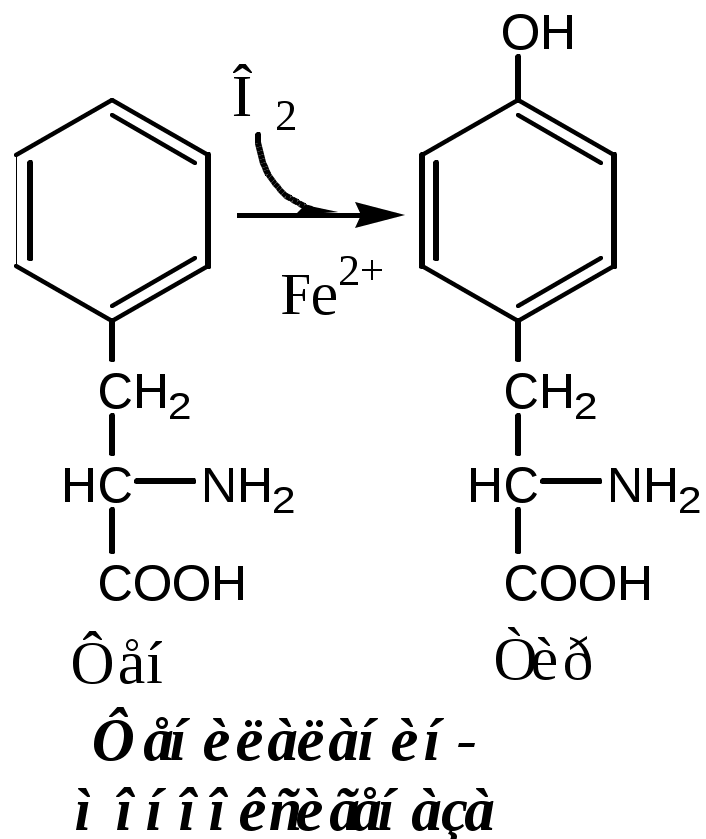
Тема: Белки III. Специфические пути обмена аминокислотФакультеты: лечебно-профилактический, медико-профилактический, педиатрический. 2 курс. ФОЛИЕВАЯ КИСЛОТА Значительную роль в обмене ряда АК, синтезе некоторых сложных липидов, нейромедиаторов, гормонов и ряда других веществ играют производные фолиевой кислоты. Фолиевая кислота широко распространёна в продуктах животного и растительного происхождения, синтезируется микрофлорой кишечника. 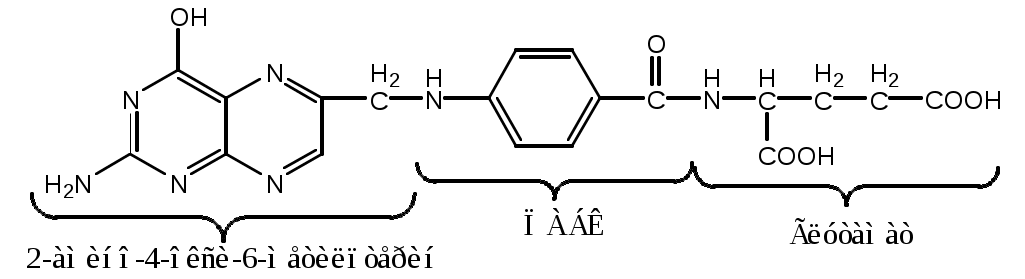 Активная форма фолиевой кислоты – ТГФК. Она образуется в печени при восстановлении фолиевой кислоты с участием фолатредуктазы и дигидрофолатредуктазы, коферментом которых служит НАДФН2. 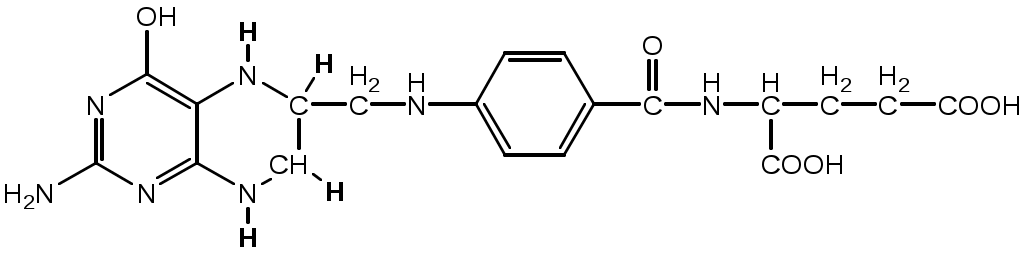 Образование одноуглеродных фрагментов, их взаимопревращения ТГФК принимает от АК одноуглеродные фрагменты: серин и глицин дают метиленовый фрагмент (-СН2-), гистидин – формимино- и формильный фрагменты. В составе ТГФК одноуглеродные фрагменты могут подвергаться взаимопревращениям: метиленовая группа превращаться в метенильную (-СН=), формильную (-НС=О), метильную (-СН3) и формиминогруппу (-CH=NH). 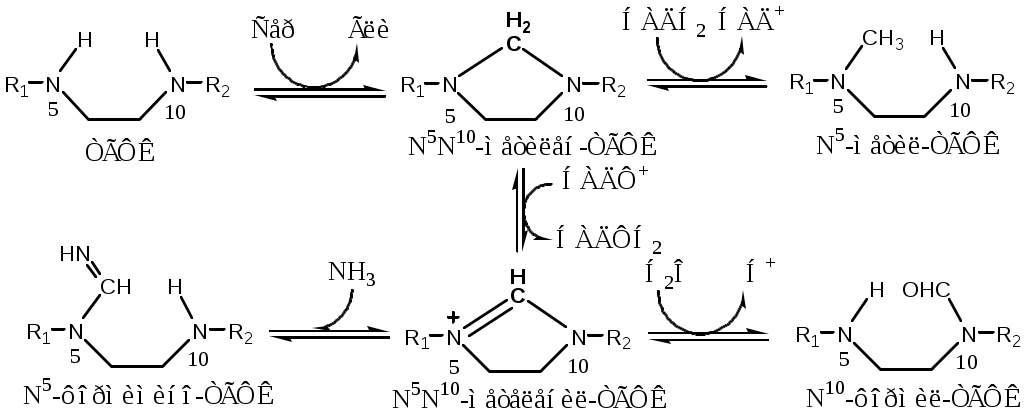 Затем ТГФК отдает одноуглеродные фрагменты на синтез новых соединений: пуриновых оснований тимидиловой кислоты регенерации метионина и т.д. Недостаточность фолиевой кислоты Гиповитаминоз фолиевой кислоты возникает редко, он приводит к: мегалобластической (макроцитарной) анемия. Она характеризуется уменьшением количества эритроцитов, снижением содержания в них гемоглобина, что вызывает увеличение размера эритроцитов. Причина — нарушение синтеза ДНК и РНК из-за недостатка тимидиловой кислоты и пуриновых нуклеотидов. лейкопении; задержке роста. КОБАЛАМИН (В12) В12 синтезируется только микроорганизмами, им богаты печень, почки. Активные формы кобаламина – метилкобаламин (цитоплазма) и дезоксиаденозилкобаламин (митохондрии). Кобаламин участвует: в передачи метила с метил-ТГФК на гомоцистеин при регенерации метионина. в превращениях одноуглеродных фрагментов в составе ТГФК. в метаболизме жирных кислот с нечетным числом атомов С и аминокислот с разветвленной цепью. Перенос протонов в реакциях изомеризации. Недостаточность В12 Гиповитаминоз возникает при нарушении всасывании В12 (дефицит фактора Касла при пониженной кислотности желудочного сока). Гиповитаминоз В12 сопровождается: макроцитарной (мегалобластической) анемией: снижение числа эритроцитов, гемоглобина, увеличение размера эритроцитов. Причина — нарушение синтеза ДНК. расстройствами деятельности нервной системы. При распаде жирных кислот с нечетным количеством атомов С и разветвленных АК из-за дефицита В12 накапливается нейротоксичная метилмалоновая кислота. ОБМЕН СЕРИНА И ГЛИЦИНА Серин и глицин - заменимые аминокислоты. Синтез серина: 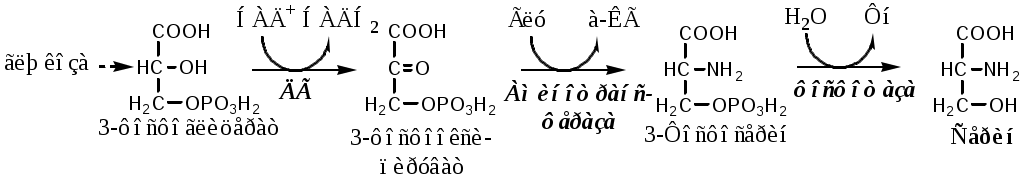 Обмен глицина: Основной путь синтеза 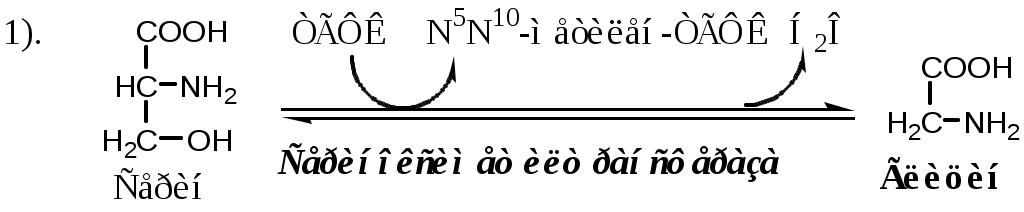 Основной путь катаболизма (в митохондриях печени)  Схема путей обмена серина и глицина Серии и глицин выполняют в организме человека разнообразные и очень важные функции. 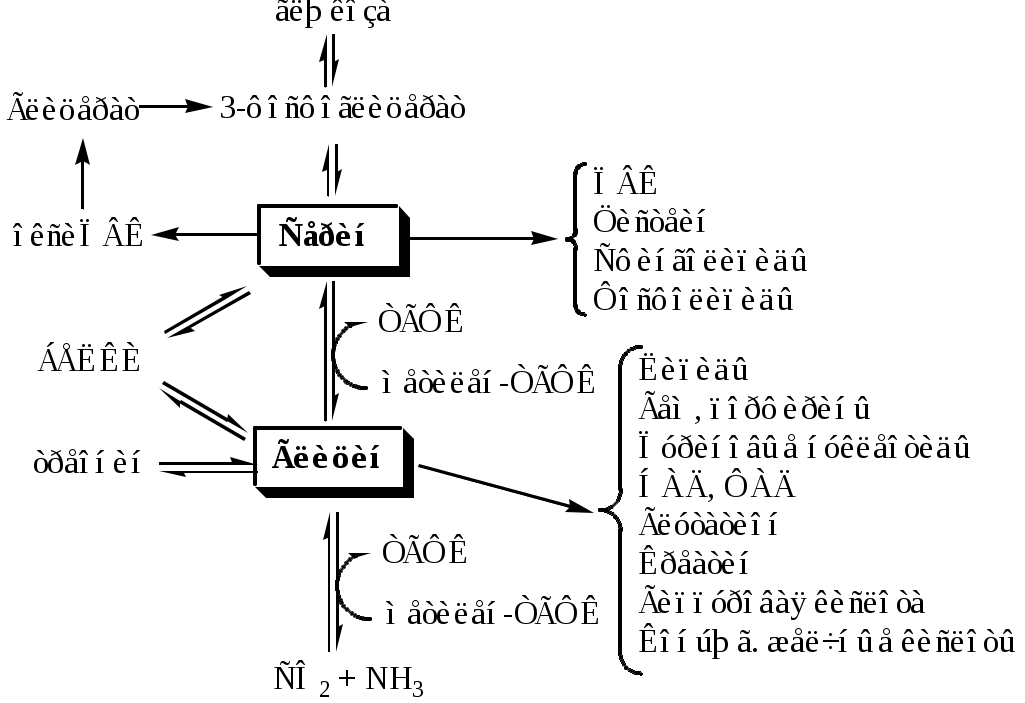 Глицин — важнейший (после ГАМК) тормозной нейромедиатор в спинном мозге, промежуточном мозге и некоторых отделах головного мозга. Наследственные нарушения обмена глицина Известно несколько заболеваний, связанных с нарушениями обмена глицина. В их основе лежит недостаточность ферментов или дефект системы транспорта этой АК. Гиперглицинемия возникает при дефекте глицинрасщепляющей системы. Проявляется повреждением мозга, судорогами, гипотонией, нарушением дыхания. Глицинурия характеризуется повышенным выделением глицина с мочой (до 1 г/сут) при нормальном содержании его в крови. Причиной является нарушение реабсорбции глицина в почках. Первичная гипероксалатурия характеризуется постоянно высоким выделением оксалата с мочой, независимо от поступления его с пищей. Дефект глицинаминотрансферазы блокирует превращение глиоксилата снова в глицин. Глицин → глиоксилат → оксалат Прогрессирует двустороннее образование оксалатных камней в мочевыводящих путях, развиваются нефрокальциноз и инфекция мочевыводящих путей. Больные погибают в детском возрасте от почечной недостаточности или гипертонии. В состав белков человека входят 2 АК, содержащие серу, — метионин и цистеин. Эти аминокислоты метаболически тесно связаны между собой. МЕТИОНИН Метионин — незаменимая аминокислота, может регенерировать из гомоцистеина с участием серина и глицина. Метионин: участвует в синтезе белков организма; является источником метильной группы, используемой в реакциях трансметилирования; является источником атома серы, необходимого для синтеза цистеина; участвует в реакциях дезаминирования; Метионил-тРНК участвует в инициации процесса трансляции. Образование S-аденозилметионина Метильная группа в метионине прочно связана с серой, поэтому донором этого одноуглеродного фрагмента служит активная форма метионина - S-аденозилметионин (SAM). (SAM — нестабилен т.к. сера при валентности 2 имеет 3 связи). SAM образуется при присоединении метионина к аденозину с участием метионинаденозилтрансферазы (есть во всех типах клеток). Аденозин образуется при гидролизе АТФ. Ресинтез метионина, роль ТГФК и витамина В12. Связь обменов метионина и цистеина 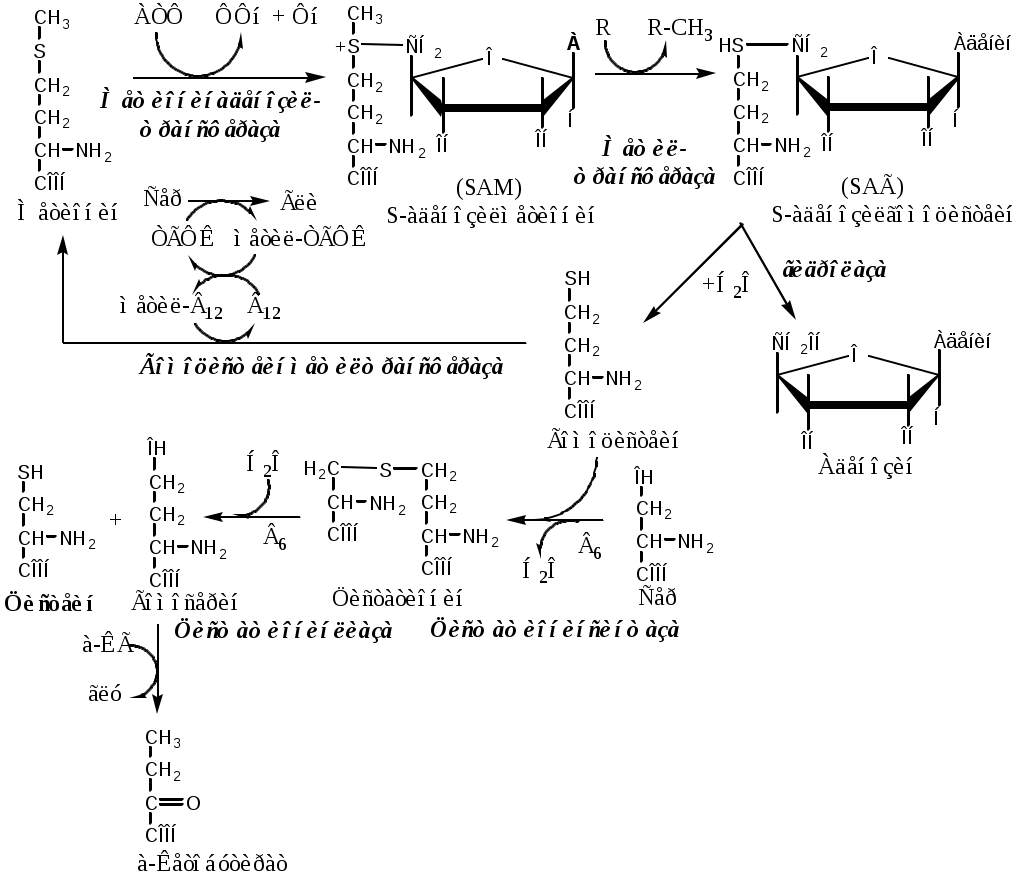 Реакции трансметилирования с участием S-аденозилметионина Реакции трансметилирования с участием S-аденозилметионинаОтщепление метильной группы от SAM и перенос её на соединение-акцептор катализируют ферменты метилтрансферазы. SAM в ходе реакции превращается в S-аденозилгомоцистеин (SAT). Синтез холина 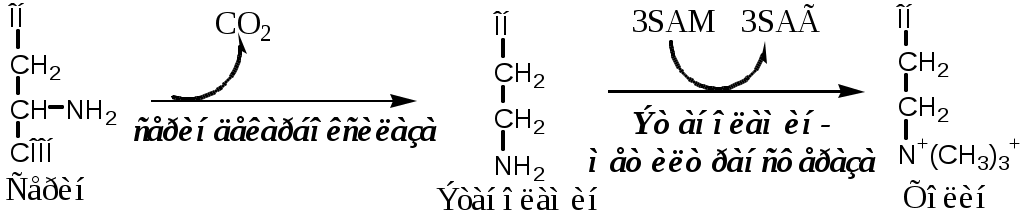 Синтез лецитина  Аналогично синтезируются: из ГАМК → карнитин; из гуанидинацетата → креатин; из норадреналина → адреналин; из карнозина → анзерин; Реакции трансметилирования используются также в синтезе азотистых оснований, инактивации гормонов, нейромедиаторов и обезвреживании ксенобиотиков. ЦИСТЕИН Цистеин – серосодержащая условнозаменимая АК. Синтезируется из незаменимого метионина и заменимого серина. Нарушение синтеза цистеина возникает при гиповитаминозе фолиевой кислоты, В6, В12 или наследственных дефектах цистатионинсинтазы и цистатионинлиазы. Гомоцистеин превращается в гомоцистин, который накапливается в крови, тканях и выделяется с мочой. Обмен цистеина: схема путей, их значение. Цистеин: используется в белках для формирования третичной структуры (дисульфидные мостики); SH группы цистеина формируют активный центр многих ферментов; идет на синтез глутатиона, таурина (парные желчные кислоты), НS-КоА, ПВК (глюкоза); Является источником сульфатов, которые идут на синтез ФАФС или выделяются с мочой. Образование сульфат-иона, его утилизация (образование ФАФС). 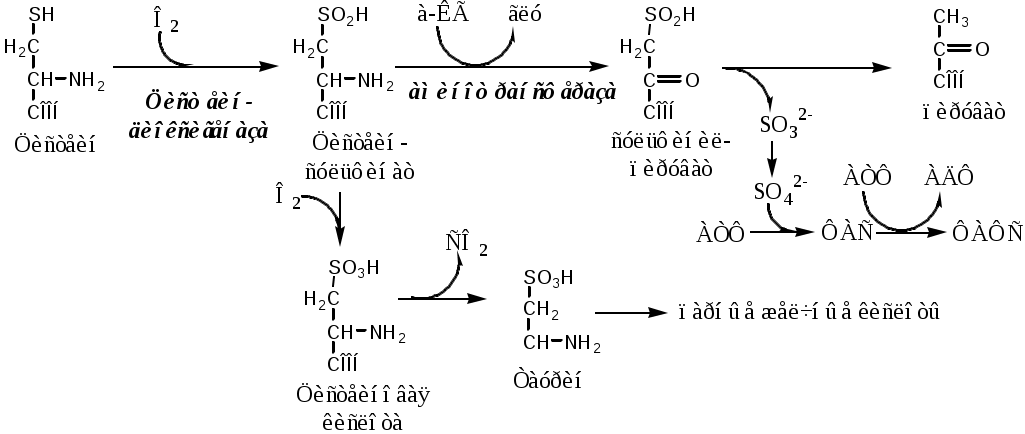 ФАФС используется: ФАФС используется:1. В обезвреживании ксенобиотиков: 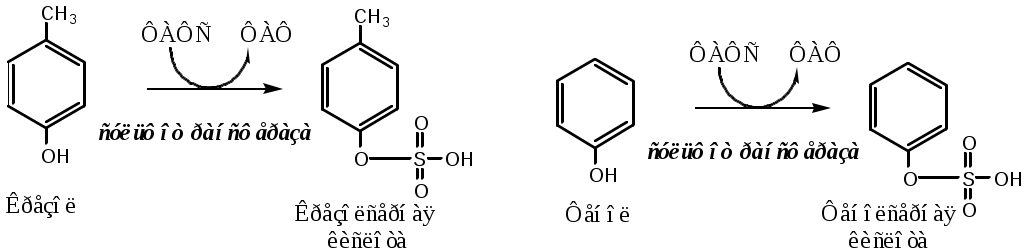 2. В синтезе гликозаминогликанов (сульфирование ОН групп производных глюкозы, галактозы сульфотрансферазой). ФЕНИЛАЛАНИН Фенилаланин — незаменимая АК, которая содержится в достаточных количествах в пищевых продуктах. Фенилаланин идет в основном на синтез белков и тирозина.
Фенилкетонурия В печени здоровых людей небольшая часть фенилаланина (10%) превращается в фениллактат и фенилацетилглутамин. При дефекте фенилаланингидроксилазы этот путь катаболизма фенилаланина становится главным, что способствует развитию фенилкетонурии (ФКУ). Классическая ФКУ — наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы (частота 1:10000 новорождённых), которые приводят к снижению активности фермента или полной его инактивации. При ФКУ концентрация фен повышается в крови в 20—30 раз, в моче — в 100—300 раз по сравнению с нормой. В крови и моче повышается содержание метаболитов альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглутамина. Проявления ФКУ: нарушение умственного и физического развития; судорожный синдром; нарушение пигментации. Проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие концентрации фенилаланина ограничивают транспорт тирозина и триптофана через гематоэнцефалический барьер и тормозят синтез нейромедиаторов (дофамина, норадреналина, серотонина). Прогрессирующее нарушение умственного и физического развития у детей, больных ФКУ, можно предотвратить диетой с очень низким содержанием или полным исключением фенилаланина. Если такое лечение начато сразу после рождения ребёнка, то повреждение мозга предотвращается. Считается, что ограничения в питании могут быть ослаблены после 10-летнего возраста (окончание процессов миелинизации мозга), однако в настоящее время многие педиатры склоняются в сторону «пожизненной диеты». При отсутствии лечения больные не доживают до 30 лет. Для диагностики ФКУ используют качественные и количественные методы обнаружения патологических метаболитов в моче, определение концентрации фенилаланина в крови и моче. Дефектный ген, ответственный за фенилкетонурию, можно обнаружить у фенотипически нормальных гетерозиготных носителей с помощью теста толерантности к фенилаланину. 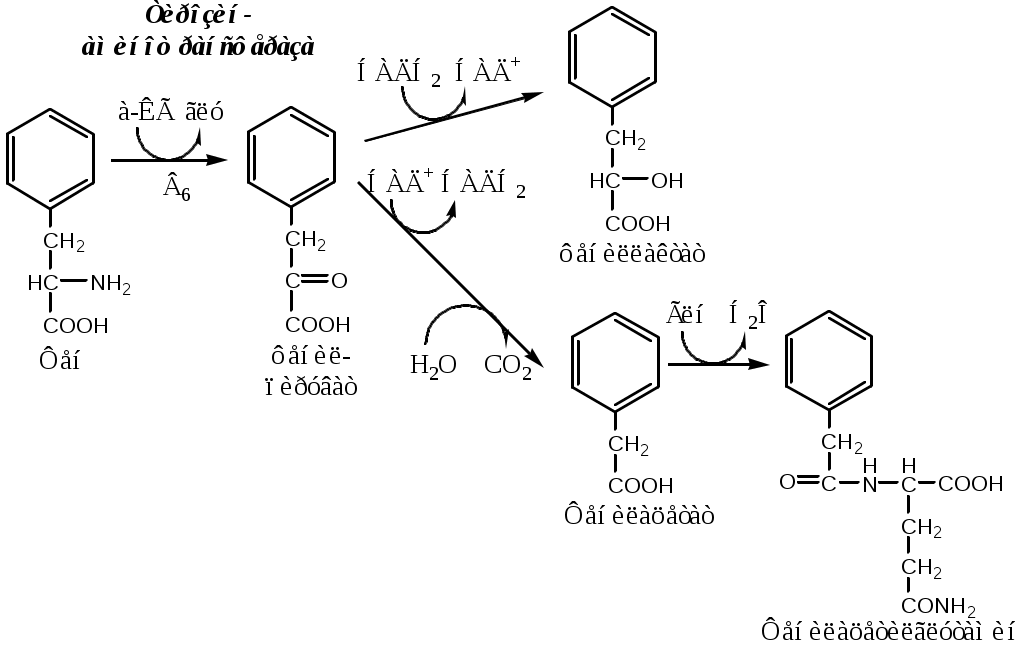 ТИРОЗИН Тирозин — условно заменимая АК, образуется из незаменимого фенилаланина. Содержание тир в пищевых белках достаточно велико. Тирозин используется в синтезе белков, катехоламинов, тиреоидных гормонов и меланинов. Обмен тирозина зависит от типа тканей. 1. Обмен тирозина в надпочечниках и нервной ткани В мозговом веществе надпочечников и нервной ткани тирозин метаболизирует по катехоламиновому пути с образованием дофамина, норадреналина и адреналина (только в надпочечниках). 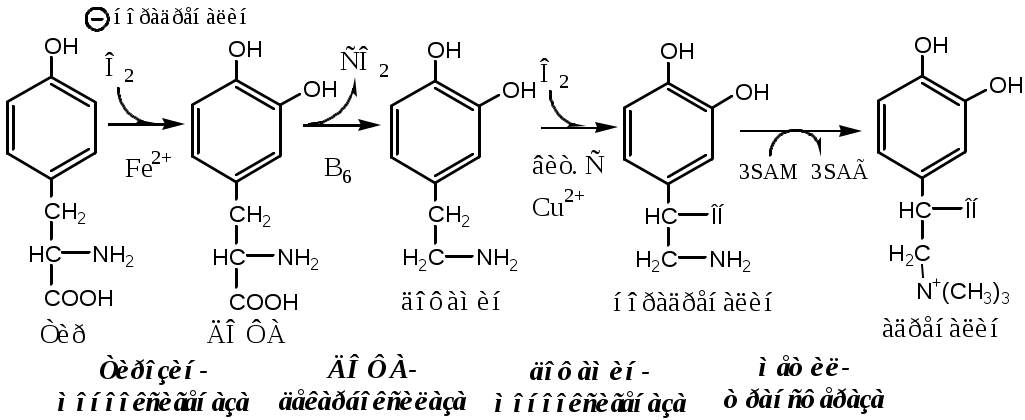 Тирозингидроксилаза (тирозинмонооксигеназа) Fe2+ -зависимый фермент, в качестве кофермента использующий Н4БП. Ее ингибирует норадреналин. Дофамин и норадреналин служат медиаторами в синаптической передаче нервных импульсов, а адреналин — гормон широкого спектра действия, регулирующий энергетический обмен. Одна из функций катехоламинов — регуляция деятельности ССС. Нарушение синтеза катехоламинов может вызывать различные нервно-психические заболевания, причём патологические отклонения наблюдаются как при снижении, так и при увеличении их количества. Снижение в нервных клетках содержания дофамина и норадреналина часто приводит к депрессивным состояниям. При шизофрении в височной доле мозга наблюдается гиперсекреция дофамина. Болезнь Паркинсона Болезнь Паркинсона развивается при снижении активности тирозинмонооксигеназы и ДОФА-декарбоксилазы, что приводит к недостаточности дофамина в чёрной субстанции мозга. Это одно из самых распространённых неврологических заболеваний (частота 1:200 среди людей старше 60 лет). Заболевание сопровождается акинезией (скованность движений), ригидностью (напряжение мышц) и тремором (непроизвольное дрожание). Дофамин не проникает через гематоэнцефалический барьер и как лекарственный препарат не используется. Для лечения паркинсонизма используют заместительную терапию препаратами-предшественниками дофамина (производными ДОФА) — леводопа, мадопар, наком и др. Также подавляют инактивацию дофамина ингибиторами МАО (депренил, ниаламид, пиразидол и др.). 2. Обмен тирозина в меланоцитах В пигментных клетках (меланоцитах) обмен тирозин идет по меланиновому пути. Из тирозина синтезируются пигменты — меланины 2 типов: эумеланины и феомеланины. Эумеланины (чёрного и коричневого цвета) — нерастворимые высокомолекулярные полимеры 5,6-дигидроксииндола. Феомеланины — жёлтые или красновато-коричневые полимеры, растворимые в разбавленных щелочах. Меланины присутствуют в сетчатке глаз, в составе волос, в коже. Цвет кожи зависит от распределения меланоцитов и количества в них разных типов меланинов. 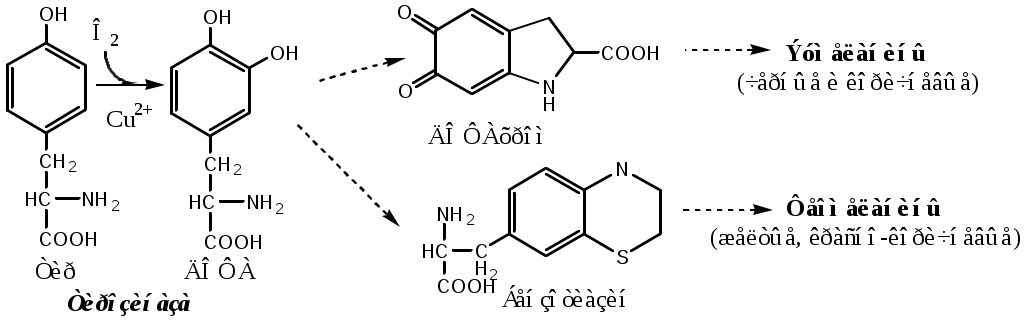 Альбинизм При наследственном дефекте тирозиназы (1:20000) в меланоцитах нарушается синтез меланинов и развивается альбинизм. Клиническое проявление альбинизма (от лат. albus — белый) — отсутствие пигментации кожи, сетчатки глаз и волос. У больных часто снижена острота зрения, возникает светобоязнь. Длительное пребывание таких больных под открытым солнцем приводит к раку кожи. 3. Превращение тирозина в щитовидной железе В щитовидной железе из тирозина синтезируются и выделяются гормоны йодтиронины: тироксин (тетрайодтиронин) и трийодтиронин. 5. Катаболизм тирозина в печени Катаболизм тирозина происходит в печени по гомогентизиновому пути (схема). 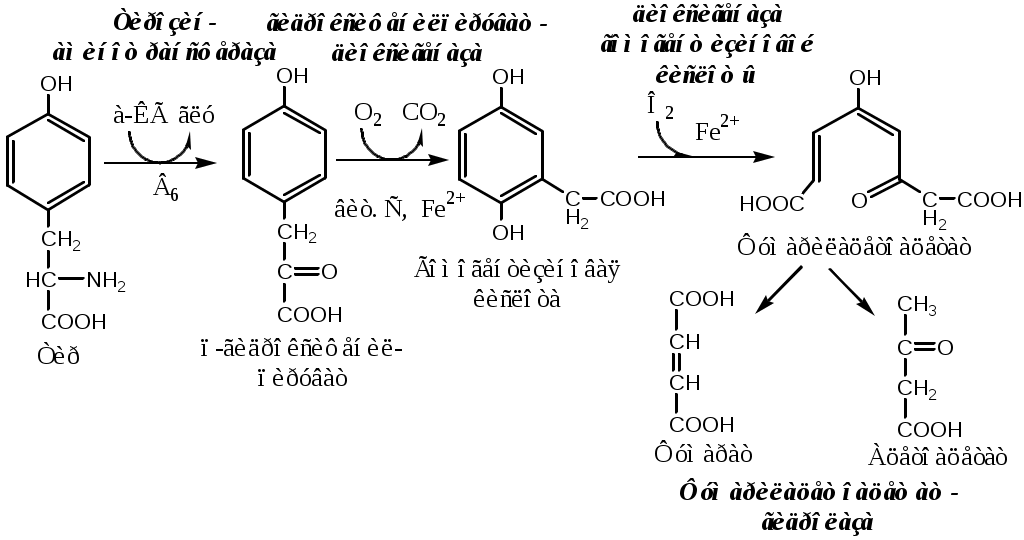 Фумарат может окисляться до СО2 и Н2О или использоваться для глюконеогенеза. Ацетоацетат — кетоновое тело, окисляемое до СО2 и Н2О с выделением энергии. Алкаптонурия («чёрная моча») При наследственном дефекте диоксигеназы гомогентизиновой кислоты (2—5 случаев на 1 млн новорождённых) развивается алкаптонурия. При алкаптонурии происходит накопление в организме гомогентизиновой кислоты, избытки которой выделяются с мочой. На воздухе гомогентизиновая кислота окисляется с образованием тёмных пигментов - алкаптонов. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Тирозинемии Некоторые нарушения катаболизма тирозина в печени приводят к тирозинемии и тирозинурии. Различают 3 типа тирозинемии. 1. Тирозинемия типа 1 (тирозиноз). Причиной заболевания является дефект фумарилацетоацетатгидролазы. Накапливающиеся метаболиты снижают активность некоторых ферментов и транспортных систем аминокислот. Патофизиология этого нарушения достаточно сложна. Острая форма тирозиноза характерна для новорождённых. Клинические проявления — диарея, рвота, задержка в развитии. Без лечения дети погибают в возрасте 6—8 мес из-за развивающейся недостаточности печени. Хроническая форма характеризуется сходными, но менее выраженными симптомами. Гибель наступает в возрасте 10 лет. Содержание тирозина в крови у больных в несколько раз превышает норму. Для лечения используют диету с пониженным содержанием тирозина и фенилаланина. 2. Тирозинемия типа II (синдром Рихнера—Ханхорта). Причина — дефект тирозинаминотрансферазы. Концентрация тирозина в крови больных повышена. Для заболевания характерны поражения глаз и кожи, умеренная умственная отсталость, нарушение координации движений. 3. Тирозинемия новорождённых (кратковременная). Заболевание возникает в результате снижения активности фермента п-гидроксифенилпируватдиоксигеназы. В результате в крови больных повышается концентрация п-гидроксифенилацетата, тирозина и фенилаланина. При лечении назначают бедную белком диету и витамин С. |