Пособие .генеологический метод. Лагкуева Фатима Катабиновна
 Скачать 1.22 Mb. Скачать 1.22 Mb.
|

|
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ СЕВЕРО-ОСЕТИНСКАЯ ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ КАФЕДРА ПСИХИАТРИИ С КУРСАМИ НЕЙРОХИРУРГИИ И МЕДИЦИНСКОЙ ГЕНЕТИКИ Лагкуева Фатима КатабиновнаГЕНЕАЛОГИЧЕСКИЙ МЕТОД В ДИАГНОСТИКЕ И ПРОФИЛАКТИКЕ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ Учебно-методические рекомендации для студентов лечебного и педиатрического факультетов, интернов, клинических ординаторов и врачей-клиницистов Владикавказ – 2005 Методические рекомендации по применению генеалогического метода в диагностике и профилактике наследственных болезней составлены доцентом курса медицинской генетики, к.м.н. Лагкуевой Ф.К. (зав.кафедрой психиатрии с курсами нейрохирургии и медицинской генетики – д.м.н., профессор Букановская Т.И.) Методические рекомендации посвящены вопросам применения генеалогического метода в диагностике и профилактике наследственных болезней. Позволяют освоить принципы и методику составления и анализа родословных с различными типами наследования нормальных и патологических признаков у человека (пробанда), а также производить расчеты риска их наследования. Методические рекомендации предназначены для студентов лечебного, педиатрического и медико-профилактического факультетов, субординаторов, интернов, клинических ординаторов и врачей различных специальностей. Рецензенты: Руководитель Центра информационной поддержки Федерального генетического регистра МНИИПДХ - ст.науч.сотр., к.м.н. Н.С.Демикова Заведующая кафедрой биологии СОГМА - профессор Л.В.Бибаева ГЕНЕАЛОГИЧЕСКИЙ МЕТОД I. Показания для применения генеалогического метода Генеалогический анализ, как метод диагностики наследственных болезней, опирается на генеалогию – учение о родословных. Суть этого метода заключается в составлении родословной и последующем ее анализе. Впервые такой подход был внедрен в медицину в 1865 г. английским врачом Френсисом Гальтоном. В нашей стране наиболее полно и широко генеалогический метод был применен в клинической практике известным клиницистом, генетиком и неврологом С.Н. Давиденковым. Генеалогический метод широко используется для решения как научных, так и прикладных проблем. Он позволяет выявить наследственный характер признака и определить тип его наследования, установить сцепление и характер взаимодействия генов, определить экспрессивность и пенетрантность аллелей, установить круг лиц, нуждающихся в детальных исследованиях для выявления носительства патологических аллелей, и производить расчеты рисков при медико-генетическом консультировании. Показаниями для применения генеалогического метода в клинической медицине могут быть:
Генеалогический метод включает два этапа: составление родословной и собственно генеалогический анализ. II. Методика и порядок составления родословной Родословная отличается от генеалогического древа тем, что помимо характера родственных связей между членами семьи, отражает информацию о проявлении какого-либо признака, состоянии здоровья или патологии среди родственников. Для построения родословной, прежде всего, необходимо собрать максимально подробную семейную информацию относительно интересующего вопроса. Сбор генеалогической информации о наличии среди родственников консультируемого тех или иных заболеваний может проводиться разными методами: методом опроса, очного или заочного анкетирования, на основе личного обследования членов семьи. Обычно родословная собирается по одному или нескольким интересующим исследователя признакам или болезням. При сборе семейных анамнестических данных могут оказаться полезными некоторые практические рекомендации. Сбор семейной информации осуществлять по определенной системе: начинать от пробанда, далее переходить к его сибсам (родным братьям и сестрам), затем - к родственникам по линии матери (ее родители, братья и сестры, племянники и племянницы) и в той же последовательности - к родственникам по линии отца. Аналогичным образом собирать информацию о родственниках по линии супруга и потомстве с указанием исходов всех состоявшихся беременностей. Такой способ сбора информации позволяет пополнить родословную сведениями по восходящему, нисходящему и боковым направлениям и получить информацию о 3-4 поколениях родственников. При сборе информации о родственниках пробанда следует акцентировать внимание не только на характере имеюшейся у них патологии (врожденной, наследственной, ненаследственной), но и на таких сведениях, как этническая и религиозная принадлежность, проживание на географически изолированной или экологически неблагополучной территории, наличие и характер профессиональных вредностей, наличие «необычных», но не нарушающих жизнедеятельность черт фенотипа, наличие случаев необоснованного отказа от вступления в брак и бесплодных браков, возраст и причина наступления смерти, особенно при редких обстоятельствах. Эффективность генеалогического анализа зависит от достоверности, правдивости и полноты полученной информации. Безусловно, важно добиться доверительных отношений между врачом и консультируемым, но все же следует помнить о том, что из-за чувства ложного стыда или желания переложить вину на другого, люди могут скрывать случаи патологии в своей семье и находить их в семье супруга. Иногда целесообразно проводить отдельно опрос каждого члена семьи, в том числе и членов супружеской пары. Для получения более точных семейных сведений можно применять анкетирование членов семьи, использовать информацию медицинских и генеалогических документов (выписки из историй болезни, медицинские заключения, домовые книги и т.д.), проводить личный осмотр и обследование родственников больного. После сбора генеалогической информации о конкретной семье, врач-генетик (или медицинская сестра) приступает к графическому изображению родословной. При построении родословной (генеалогического древа) используют определенные общепринятые принципы и символы, применение которых позволяет четко и единообразно толковать генеалогическую информацию о конкретной семье. Принципы составления родословной:
Символы, используемые при составлении родословной 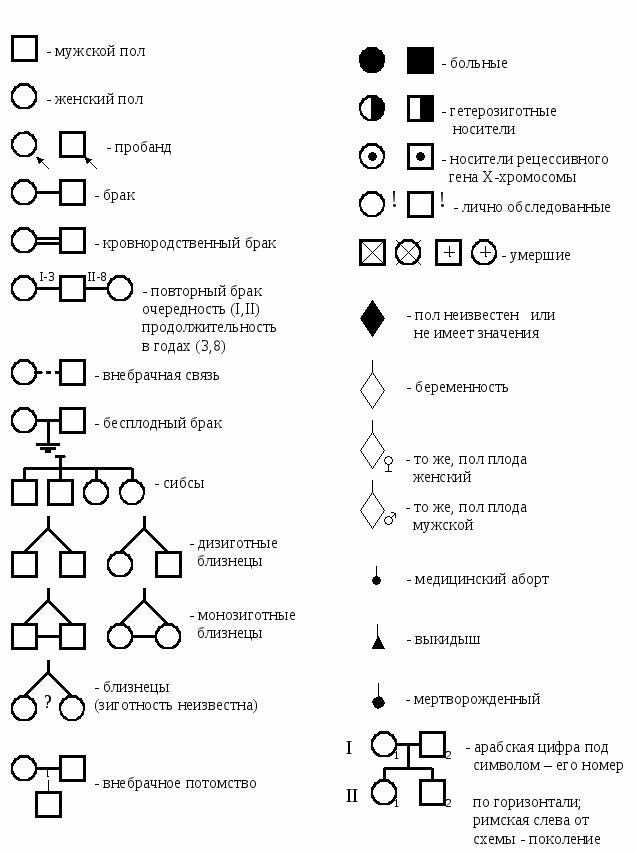 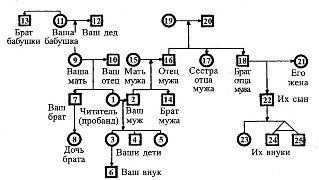 Рис. 1. Примерная схема (порядок) составления родословной 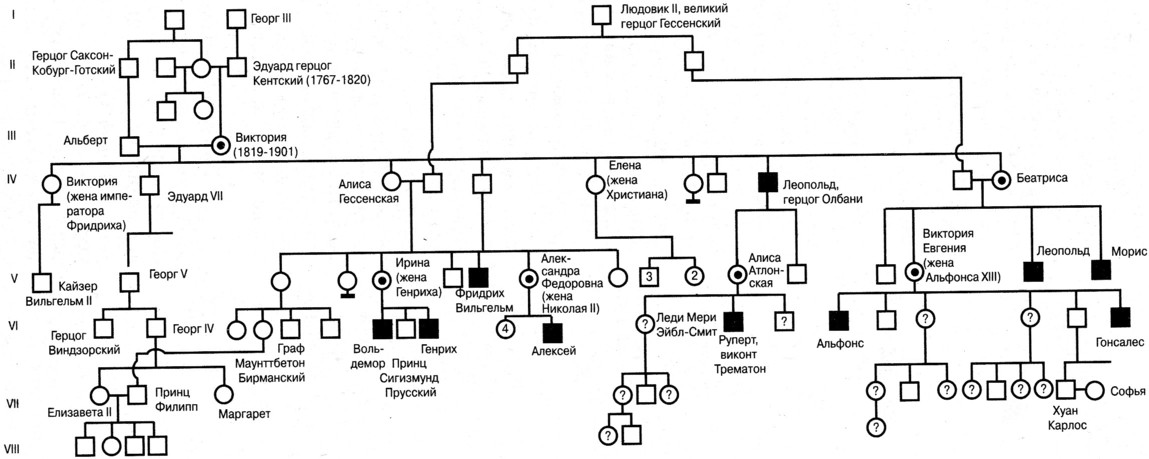 Рис. 2. Пример генеалогического древа (родословной) Европейских королевских семей (наследование гемофилии). III. Генеалогический анализ и особенности родословных с разными типами наследования При проведении анализа родословной, прежде всего, следует ответить на вопрос: является ли анализируемый признак (или заболевание) в семье единичным или же встречается несколько раз? В последнем случае необходимо исключить возможность фенокопии или семейной патологии ненаследственного генеза и предположить наследственный характер признака. Далее приступают к установлению типа наследования, для чего следует вспомнить основные генетические закономерности и изучить характерные черты родословных с разными типами наследования. Аутосомно-доминантное наследование – наследование признаков (или болезней), развитие которых детерминируется доминантным аллелем (А) гена, локализованного в одной из аутосом. В этом случае нормальный вариант признака (или отсутствие болезни) определяется рецессивным (а) аллелем того же гена. В соответствие с менделевскими закономерностями доминантный аллель, как правило, полностью (при полном доминировании) подавляет действие рецессивного аллеля, а потому признак (или болезнь) проявляется как у доминантных гомозигот (АА), так и у гетерозигот (Аа), и не проявляется у рецессивных гомозигот (аа). Таким образом, больные или обладающие анализируемым признаком члены родословной будут иметь генотип АА или Аа, а здоровые и не обладающие таким признаком – генотип аа. В связи с тем, что многие доминантные гены в гомозиготном состоянии оказываются летальными, наиболее частыми типами браков в популяции оказываются браки между больными (Аа Аа) или между больными и здоровыми (Аа аа). Родословная с аутосомно-доминантным типом наследования имеет следующие характерные черты:
Пенетрантность – это вероятность проявления гена. Она выражается в процентах заболевших от числа носителей гена. Так, если доминантный ген проявляется в фенотипе у всех его обладателей, то его пенетрантность равна 100%. Если среди носителей патологического доминантного гена болезнь проявляется только у половины, то пенетрантность равна 50%. Разные гены обладают различной пенетрантностью: ген ретинобластомы – 80%, ген отосклероза – 40%. Носительство доминантного гена без фенотипического проявления можно заподозрить у одного из родителей, если болезнь проявилась более чем у одного из его детей, или если он имеет больных родственников (сибсов, родителей). Доминантные гены обладают и различной экспрессивностью (степенью выраженности действия гена), что обуславливает различную степень тяжести заболевания даже в пределах одной семьи – от легких и стертых форм до выраженных и крайне тяжелых. Это отчасти объясняется неполным подавлением рецессивного аллеля (гетерозиготы имеют среднюю степень выраженности доминантного признака) или влиянием других генов в генотипе. Аутосомно-доминантно у человека наследуются: резус-антиген на мембране эритроцитов, пигмент в волосах, коже и радужной оболочке глаза. К числу заболеваний с аутосомно-доминантным типом наследования относятся: ахондроплазия, несовершенный остеогенез, синдром Марфана, семейная гиперхолестеринемия, болезнь Виллебрандта, хорея Гентингтона, туберозный склероз, нейрофиброматоз Реклингаузена и др. 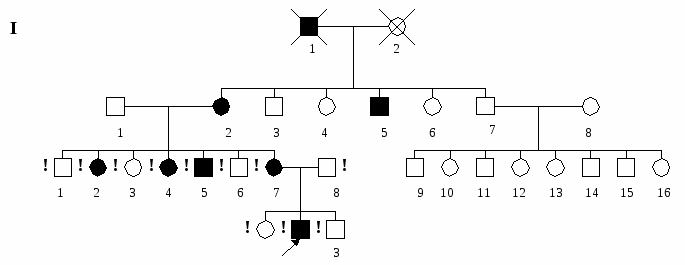 III 2 1 IV II Рис. 3. Родословная семьи с аутосомно-доминантным наследованием болезни (синдром Марфана). Аутосомно-рецессивное наследование - наследование признаков (или болезней), развитие которых детерминируется рецессивным аллелем (а) гена, локализованного в одной из аутосом. В этом случае нормальный вариант признака (или отсутствие болезни) определяется доминантным (А) аллелем того же гена. Рецессивный аллель проявляется только в отсутствие доминантного аллеля, т.е. только у гомозигот (аа). Поэтому в гетерозиготном состоянии (Аа) он может существовать во многих поколениях, никак не проявляясь фенотипически и накапливаясь в семье. В результате первый больной рецессивной патологией появляется в семье через многие поколения после возникновения мутации. Фенотипически (или клинически) здоровые люди могут оказаться как доминантными гомозиготами (АА), так и гетерозиготными носителями патологического аллеля (Аа). Наиболее типичным типом брака при аутосомно-рецессивном наследовании является брак между двумя гетерозиготными носителями – АаАа, реже наблюдаются браки между больными и здоровыми типа ааАА или ааАа. Родословная с аутосомно-доминантным типом наследования имеет следующие характерные черты:
Частота аутосомно-рецессивной патологии возрастает в изолятах и популяциях, где существует высокий процент кровнородственных браков. Это объясняется тем, что в семьях, отягощенных рецессивным геном, концентрация гетерозиготных носителей выше, чем в общей популяции, а вероятность рождения больного ребенка оказывается тем выше, чем выше степень кровного родства между супругами и процент общих генов. Таблица 1. Доля общих генов у родственников разной степени родства
Значительно реже рождение больного с аутосомно-рецессивной патологией можно объяснить гетерозиготностью одного родителя и случайно возникшей аналогичной мутацией в гамете второго - здорового родителя. Чтобы правильно оценить величину повторного риска в семье, необходимо установить гетерозиготность родителей по аутосомно-рецессивному гену. Аутосомно-рецессивно у человека наследуются: низкий рост, голубые глаза, резус-отрицательный фактор крови. К числу заболеваний с аутосомно-рецессивным типом наследования относятся: альбинизм, муковисцидоз, гомоцистинурия, фенилкетонурия, гепатолентикулярная дегенерация, атаксия Фридрейха и др. 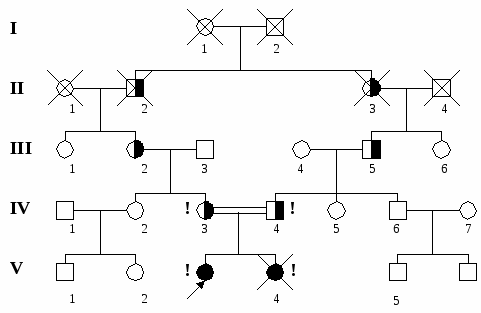 3 6 Рис. 4. Родословная семьи с аутосомно-рецессивным наследованием болезни (фенилкетонурия). Х-сцепленное наследование – наследование признаков (или болезней), гены которых локализуются в Х-хромосоме. Главными особенностями Х-сцепленного наследования являются: отсутствие передачи соответствующего гена от отца – сыну (поскольку мужчины передают свою единственную Х-хромосому только дочерям) и активность любого (доминантного или рецессивного) аллеля Х-сцепленных генов у мужчин (поскольку они, обладая одной Х-хромосомой, являются гемизиготными по этим генам). Женщины, обладающие двумя Х-хромосомами, могут быть как гомо-, так и гетерозиготными по Х-сцепленным генам, и проявление патологии у них зависит от доминантности или рецессивности патологического аллеля. Различают Х-сцепленное доминантное и Х-сцепленное рецессивное наследование. Для Х-сцепленного доминантного наследования, когда патологическим является доминантный аллель, характерны следующие признаки:
Некоторые доминантные Х-сцепленные патологические признаки оказываются летальными для мужчин (например, очаговая гиперплазия кожи, рото-лице-пальцевой синдром и гипераммониемия, вызываемая дефицитом орнитинтранскабаламиназы). В таких случаях наследование характеризуется следующими особенностями:
К числу заболеваний с Х-сцепленным доминантным наследованием относятся: фосфат-диабет (витамин-D резистентный рахит), синдром Коффина-Лоури, рото-лице-пальцевой синдром, синдром недержания пигмента. 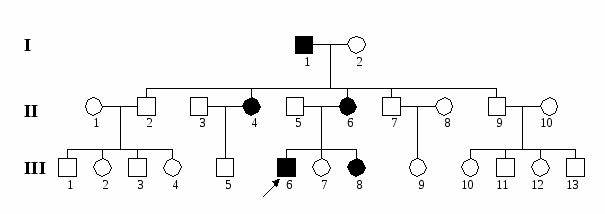 Рис. 5. Родословная семьи с Х-сцепленным доминантным наследованием болезни (витамин D-резистентный рахит). Родословные при Х-сцепленном рецессивном наследовании, когда патологический аллель гена Х-хромосомы является рецессивным, несколько различаются в зависимости от того, нарушается ли репродуктивный статус больного при данной патологии или нет. В тех случаях, когда больные оказываются бесплодными (например, при мышечной дистрофии Дюшенна-Беккера), родословные имеют следующие характерные черты:
Если репродукция при данной болезни не нарушена (например, при гемофилии), то наследование характеризуется следующим образом:
К Х-сцепленным рецессивным болезням относятся: гемофилия А и В, дальтонизм, мышечная дистрофия Дюшенна-Беккера, болезнь Фабри, синдром Хантера (мукополисахаридоз II типа), синдром тестикулярной феминизации и синдром Мартина-Белл (синдром ломкой Х-хромосомы). 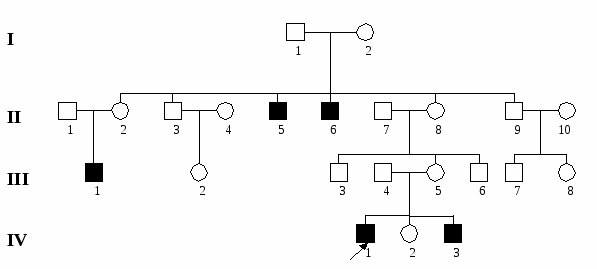 Рис. 6. Родословная семьи с Х-сцепленным рецессивным наследованием болезни с нарушением репродукции (мышечная дистрофия Дюшенна). 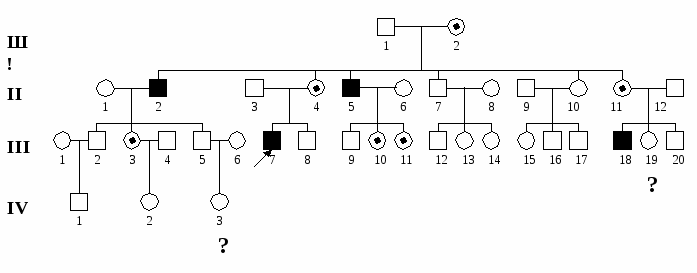 Рис. 7. Родословная семьи с Х-сцепленным рецессивным наследованием болезни без нарушения репродукции (гемофилия А). У-сцепленное наследование – наследование признаков, гены которых локализуются в эухроматиновых районах У-хромосомы. В настоящее время в У-хромосоме картировано несколько генов: ген, детерминирующий развитие семенников, фактор азооспермии, ген, определяющий оволосение ушной раковины и одну из форм ихтиоза и другие. Понятно, что говорить о наследовании мутаций, затрагивающих формирование семенников и сперматогенез, не приходится, поскольку их обладатели стерильны. В других случаях признак (или болезнь) проявляется только у мужчин, а больные отцы передают патологический аллель всем сыновьям, но не дочерям. 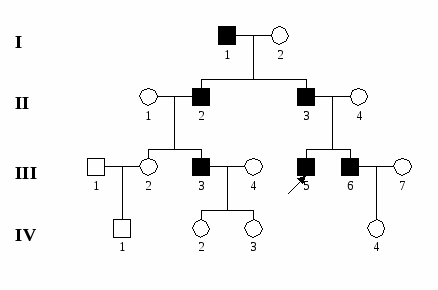 Рис. 8. Родословная семьи с У-сцепленным наследованием болезни (гипертрихоз ушной раковины). Митохондриальное, или цитоплазматическое наследование – это наследование генов, локализованных в ДНК митохондрий. Особенности этого типа наследования определяются тем, митохондрии в клетках человека всегда имеют материнское происхождение, поскольку попадают в зиготу только с цитоплазмой яйцеклетки (головка спермиев практически лишена цитоплазмы и цитоплазмтических структур). Митохондриальная ДНК содержит несколько тысяч генов. Мутации этих генов приводят к развитию довольно тяжелых заболеваний нервной, мышечной системы и органов чувств, которые составляют особую группу патологии человека - митохондриальные болезни. Для митохондриального наследования характерны следующие черты:
К митоходриальным болезням относятся: атрофия зрительного нерва Лебера, прогрессирующая офтальмоплегия, синдром Цельвегера, синдром Кернса-Сейра, митохондриальная миопатия и др. 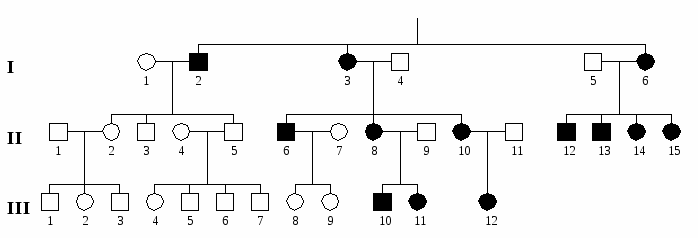 Рис. 9. Родословная, иллюстрирующая митохондриальное наследование (нейтральный признак-фрагмент мДНК). Мультифакториальное наследование – наследование признаков и болезней с наследственным предрасположением, которые имеют мультифакторную природу и являются результатом взаимодействия многих генов с многочисленными факторами внешней среды. К таким болезням человека относятся распространенные врожденные пороки развития и хронические соматические, психические и нервные болезни. Анализ родословных при мультифакториальных заболеваниях основан не на менделевских закономерностях, как при моногенных признаках и болезнях, а на эмпирических данных, полученных при анализе многих семей. Мультифакториальный тип наследования характеризуется следующими особенностями:
В таблице 2 приводятся данные о распространенности болезней с наследственной предрасположенностью. Величины эмпирического риска при разных мультифакториальных болезнях представлены в таблице 3. Там же приводятся данные по заболеваниям с неизвестным типом наследования. Таблица 2. Распространенность болезней с наследственной предрасположенностью
Таблица 3. Эмпирический риск при мультифакториальных заболеваниях и болезнях с неясным типом наследования
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
СОДЕРЖАНИЕ I. Показания для применения генеалогического метода 2 II. Методика и порядок составления родословной 2 III. Генеалогический анализ и особенности родословных с разными типами наследования 8
IV. Рекомендуемая литература 20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||