лекция идентификация веществ. Лекции Определение температуры плавления. Спектральные методы идентификации органических веществ
 Скачать 493.03 Kb. Скачать 493.03 Kb.
|

|
Лекция МЕТОДЫ ИДЕНТИФИКАЦИИ ОРГАНИЧЕСКИХ ВЕЩЕСТВ Схема лекции Определение температуры плавления. Спектральные методы идентификации органических веществ. 2.1. Применение спектроскопии ядерного магнитного резонанса в органической химии. ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫ ПЛАВЛЕНИЯ Температурой плавления вещества называют температуру, при которой твердое вещество находится в равновесии с собственным расплавом. Температура плавления является физической константой вещества и используется для его идентификации и определения степени его чистоты. Чистые вещества обладают очень резко выраженной точкой плавления. Присутствие в веществе даже незначительных загрязнений резко влияет на температуру плавления. Обычно присутствие примесей приводит к значительному снижению температуры плавления. Если вещество, являющееся примесью имеет более высокую температуру плавления, чем основное вещество, в большинстве случаев также наблюдается понижение, температуры плавления, хотя бывают исключения. Именно снижение температуры плавления вещества при наличии загрязнений используется для определения чистоты вещества. Для идентификации полученного вещества его пробу смешивают с таким же веществом в равных количествах. Затем проводят определение температуры плавления этой смеси (‘проба смешанного плавления’). Если при этом температура плавления остается неизменной, можно сделать заключение об идентичности двух смешанных веществ. Если температура плавления стает ниже (депрессия температуры плавления), то значит, вещества отличаются друг от друга. При определении температуры плавления нужно пиметь в виду, что многие органические вещества плавятся с разложением. Обычно это обнаруживается по появлению или изменению окраски или по выделению газа. Температура разложения вещества не так резко выражена, как температура плавления, часто зависит от скорости нагревания. По этой причине эксперименты по определению температуры разложения не всегда воспроизводятся. Существует определенная зависимость между температурой плавления вещества и его молекулярным строением. Например, вещества с симметричными молекулами плавятся при более высокой температуре, чем вещества с менее симметричной структурой. Так, например температура правления н-бутана составляет -138,40С, а температура плавления изобутана - -159,60С; температура плавления н-пентана составляет -1300С, а температура плавления изопентана - -1600С. У геометрических изомеров транс-изомер, как правило, плавится при более высокой температуре. Так малеиновая кислота (цис-изомер) имеет температуру плавления 1300С, а транс-изомер того же вещества, фумаровая кислота, плавится при 2870С. Температуру плавления вещества чаще всего определяют в капилляре. Для этого предварительно высушенное и измельченное вещество помещают в запаянный с одного конца капилляр слоем толщиной 5 мм. Затем капилляр с веществом помещается в прибор для определения температуры плавления таким образом, чтобы вещество находилось как можно ближе к шарику термометра. В качестве теплоносителя в приборе используется серная кислота. При использовании серной кислоты в качестве теплоносителя можно определять температуры плавления веществ до 240-2500С. При более высоких температурах серная кислота начинает разлагаться.  Рисунок 1. Прибор для определения температуры плавления. Первоначально проводят измерение ориентировочной температуры плавления. При этом прибор нагревают со скоростью 8-100 в минуту и наблюдают, при какой температуре вещество плавится. Однако, быстрыое нагревание прибора не позволяет точно зафиксировать температуру плавления. По этой причине после ориентировочного определения проводят точное определение. При этом прибор охлаждают до температуры на15-20 градусов ниже чем полученная ориентировочная температура плавления и повторяют эксперимент. Теперь прибор нагревают со скоростью 1-2 градуса в минуту и точно фиксируют температуру плавления. Если вещество плавится не сразу, Что обычно бывает, то фиксируют температуру начала плавления, когда в капилляре только появляется жидкая фаза, и температуру конца плавления, когда вся масса вещества становится жидкой. Чем меньше разница между этими температурами, тем чище вещество. Опыт повторяют еще два раза и рассчитывают средний результат. В случаях, когда вещество имеет высокую температуру плавления, для ее определения используют металлический блок, в который помещают термометр с прикрепленным к нему капилляром с веществом. Существуют и более сложные приборы для определения температуры плавления. Одним из таких приборов является блок Кофлера, в котором кристаллы вещества помещаются на предметный столик микроскопа. Столик подогревается и при этом с помощью микроскопа наблюдают плавление вещества. СПЕКТРАЛЬНЫЕ МЕТОДЫ ИДЕНТИФИКАЦИИ ОРГАНИЧЕСКИХ ВЕЩЕСТВ В середине ХХ века в практику химиков вошли физические методы идентификации и исследования веществ. Эти методы быстро получили широкое распространение, так как обладали многими преимуществами перед классическими методами исследования. Во-первых, физические методы исследования обладают высокой чувствительностью. Для исследования требуется очень небольшое количество вещества. Во многих случаях есть возможность проводить анализ без разделения смеси. Во-вторых, сам анализ занимает очень мало времени. Наконец, Физические методы исследования дают очень ценную информацию о структуре молекул, которую нельзя получить другими способами. Среди всех физических методов для исследования органических веществ наиболее широко в настоящее время используются спектроскопия ядерного магнитного резонанса (ЯМР-спектроскопия), ультрафиолетовая спектроскопия (УФ-спектроскопия) и масс спектроскопия. Применение спектроскопии ядерного магнитного резонанса в органической химии Впервые явление ядерного магнитного резонанса наблюдали в 1946 году Пурселл и Блох. За открытие этого явления в 1952 году им была присуждена Нобелевская премия. Уже в 1953 году метод ЯМР был впервые применен для изучения структур химических соединений. Химики быстро оценили все преимущества данного метода и стали его широко использовать. Теоретические основы метода ЯМР. Любой атом состоит из положительно заряженного ядра и отрицательно заряженных электронов, вращающихся вокруг него. Характеристиками ядра являются его масса и заряд. Другой характеристикой ядра является его спиновое число, обусловленное вращением ядра вокруг собственной оси. Так как ядро заряжено, его вращение приводит к круговому движению заряда, что эквивалентно электрическому току, движущемуся в замкнутом проводнике. Вращающееся ядро создает магнитное поле и ведет себя как крохотный магнит. Его можно характеризовать дипольным моментом μ. Магнитный момент имеют не все ядра. Так 12С, 16О, 32S не имеют магнитного момента. Такие ядра как 1Н, 13С, 19F магнитны. Чтобы ядро обладало магнитным моментом, оно должно иметь нечетное значение атомной массы. Если на вращающееся ядро подействовать некоторым постоянным магнитным полем с напряженностью Н0, то это поле будет стремиться расположить магнит вдоль линий поля, но так как магнит вращается возникает прецессия вектора μ вокруг оси поля со скоростью ω. ω = γ Н0 , где γ- фактор пропорциональности, который называется гиромагнитным отношением и является константой для данного ядра.  Теперь перпендикулярно к Н0 приложим некоторое магнитное поле с напряженностью Н1. Это поле будет стремиться отклонить μ в плоскостьxy, но для того чтобы это произошло, необходимо, чтобы поле Н1 вращалось со скоростью, равной скорости прецессии ω = ω1. Если ω1 медленно изменять, то в момент, когда Скорость вращения поля Н1 станет равной скорости прецессии, угол θ резко изменится, что будет соответствовать обмену энергией между прецессирующим ядром и вращающимся полем Рис.2. Н1. Это явление и называется ядерным магнитным резонансом. В этот момент происходит переориентация магнитных ядер в поле Н0, что соответствует поглощению или испусканию излучения. Величина изменения энергии ΔЕ = hν, где h- константа Планка, а ν- частота излучения. Теперь перпендикулярно к Н0 приложим некоторое магнитное поле с напряженностью Н1. Это поле будет стремиться отклонить μ в плоскостьxy, но для того чтобы это произошло, необходимо, чтобы поле Н1 вращалось со скоростью, равной скорости прецессии ω = ω1. Если ω1 медленно изменять, то в момент, когда Скорость вращения поля Н1 станет равной скорости прецессии, угол θ резко изменится, что будет соответствовать обмену энергией между прецессирующим ядром и вращающимся полем Рис.2. Н1. Это явление и называется ядерным магнитным резонансом. В этот момент происходит переориентация магнитных ядер в поле Н0, что соответствует поглощению или испусканию излучения. Величина изменения энергии ΔЕ = hν, где h- константа Планка, а ν- частота излучения.γ Н0 ν = --------- 2π На практике оказалось удобнее варьировать напряженность магнитного поля Н0. При определенном значении Н0 энергия, необходимая для переориентации диполя становится равной энергии излучения, происходит поглощение энергии и наблюдается сигнал, фиксируемый прибором. Совокупность таких сигналов называется спектром ЯМР. Принципиальная схема ЯМР-спектрометра На рисунке 3 приведена принципиальная схема ЯМР-спектрометра. Ампула с образцом помещается в катушку L. Генератор G создает в катушке постоянное магнитное поле с напряженностью Н1. Напряженность внешнего магнитного поля Н0 постепенно увеличивается с помощью свип-генератора и.  Рис.3. Принципиальная схема ЯМР-спектрометра. дополнительных катушек. Свип-генератор подает ток на горизонтальные отклоняющие осциллографа. Когда величина Н0 достигнет критического состояния, система входит в резонанс. Поглощение энергии в катушке L вызывает сигнал в цепи генератора G, что отражается на экране осциллографа в виде пика. Наиболее часто в органической химии используются ЯМР-спектры ядер водорода – протонов, то есть протонный магнитный резонанс – ПМР. Основные параметры спектра ПМР Химический сдвиг Гиромагнитное отношение γ зависит от природы ядра атома, то есть для всех протонов величина γ одинакова. В этом случае все протоны будут резонировать при одной частоте и тогда в спектре любого органического вещества должен был бы присутствовать всего один сигнал, соответствующий резонансу протонов. В этом случае мы бы не получили никакой информации о строении вещества. К счастью, это не так. Частота, при которой проглощает протон, зависит не только от напряженности поля Н0, но и от наличия соседних протонов и электронного окружения. Их магнитные поля также влияют на резонанс протона, в результате эффективное магнитное поле, действующее на реальный протон, отличается от Н0. По этой причине протоны, имеющие различное окружение в молекуле, будут резонировать при различных значениях Н0. Наоборот, протоны, имеющие одинаковое окружение, будут резонировать при одном и том же значении Н0. Такие протоны называются эквивалентными. Число сигналов в ПМР-спектре зависит от числа групп эквивалентных протонов. Так как на протон влияет окружение, реальный протон в молекуле по сравнению с ”голым” протоном требует для резонанса поля с большей или меньшей напряженностью. Происходит “сдвиг сигнала” в сторону более сильного или более слабого поля. Смещение сигналов в спектре ПМР, вызванные окружением протона называются химическими сдвигами. Величина химического сдвига определяется характером химических связей в молекуле, типом группы, влиянием других групп и индукционными эффектами заместителей. Химический сдвиг выражается в миллионных долях общего приложенного магнитного поля (м.д.). В качестве начала отсчета обычно используется сигнал протонов тетраметилсилана (ТМС). Спектр записывают так, чтобы напряженность поля возрастала слева направо. Подавляющее большинство сигналов при этом располагается левее сигнала ТМС в области более слабых полей. В ПМР спектроскопии обычно используется шкала δ. Δν 106 δ = ---------------------------------------- рабочая частота прибора в Мгц В шкале δ сигнал ТМС принимается за 0. Реже используется шкала τ где сигнал ТМС принимается за 10, τ= 10-δ. Химические сдвиги для различных групп протонов приведены в таблице. ХИМИЧЕСКИЕ СДВИГИ ПРОТОНОВ ОТНОСИТЕЛЬНО ТМС Протонная группа | δ, м.д. | Протонная группа | δ, м.д. СН3 - R 0,8-1,2 R3CH 1,4-1,6 CH3 – C(R0=C< 1,6-1,9 R2 CH-OH 3,5-3,8 CH3 – Ar 2,2-2,5 Ar2 CH-OH 5,7-5,8 CH3 –I 2,16 R2 CH-NH2 2,8-3,0 CH3 –Br 2,62 R-OH 3-6 CH3 –Cl 3,0 Ar- OH 6-8 CH3 –OH 3,47 R-NH- 2-4 CH3 –C(O)-R 2,1-2,4 R-COOH 10-12 CH3 –C(O)-Ar 2,4-2,6 -CHCl2 5,3 CH3 –C(O)-OR 1,9-2,2 -CH=CH- 5-7 CH3 –C(O)-OAr 2,0-2,5 -CHCl3 7,3 CH3 –OR 3,2-3,5 -C(O)-H 9-10 CH3 –OAr 3,7-4,0 CH3 –O-C(O)-R 3,6-3,9 C6H5X CH3 –N< 2,2-2,6 X=H 7,2 R-CH2-R 1,1-1,5 X=OH 6,9 R-CH2-Ar 2,5-2,9 X=Cl 7,2 R-CH2-I 3,2 X=NO2 7,3-8,0 R-CH2-Br 3,3 X=NH2 6-7 R-CH2-Cl 3,4-3,7 X=CH3 7,2 R-CH2-OH 3,2-3,5 X=C2H5 7,2 R-CH2-C(O)-R 2,5-2,9 R-CH2-OAr 3,9-4,3 R-CH2-OC(O)-R 3,7-4,1 R-CH2-N< 2,4 Таким образом, по величинам химических сдвигов можно определить тип резонирующего протона. На рисунках 4-5 приведены спектры уксусной кислоты и толуола. 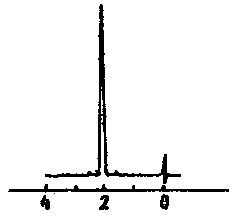 Рис.4. Спектр ПМР уксусной кислоты.  Рис. 5. Спектр ПМР толуола. 2.1.3.2.Спин-спиновое взаимодействие Каждый из вышеприведенных спектров состоит из отдельных пиков. На самом деле, чаще в ПМР-спектре наблюдаются не отдельные пики, а группы пиков. В спектрах ПМР одиночный пик называют синглетом, группа из двух пиков называется дублетом, из четырех – квартетом, из пяти – квинтетом, из шести – секстетом. В качестве примера приведем спектр бромистого этила (Рис.6). В спетре бромистого этила наблюдается два сигнала, один в виде триплета, а другой в виде дублета.  Рис.6. Спектр ПМР бромистого этила. Расщепление сигнала происходит под влиянием соседних протонов. В нашем примере на протоны группы СН3 влияет магнитное поле протоном группы СН2, а на протоны группы СН2 влияет, в свою очередь, магнитное поле протоном группы СН3. Рассмотрим как проявляется взаимное влияние магнитных полей протонов на простом примере -СН-СН2-. Поле, в котором находятся протоны группы СН2 может усиливаться или уменьшаться под влиянием магнитного поля, наведенного соседним протоном группы СН. Это поле может быть направлено или вдоль линий поля Н0 или противоположно. Таким образом, протон оказывается в двух различных магнитных полях. Взаимодействие с одним протоном дает дублет с отношением интенсивностей пиков 1:1.  В сою очередь, на протон группы СН влияют магнитные поля протонов группы СН2, что приводит к его расщеплению на три пика.  Взаимодействие с двумя протонами дает триплет с отношением интенсивностей пиков 1:2:1. В качестве примера рассмотрим спектр 2-бромпропана, приведенный на рисунке 7. В общем случае, мультиплетность сигнала равна n+1, где n – число соседних протонов.  Рис.7. Спектр ПМР 1-бромпропана. Протоны группы СН3 дают сигнал в области 1,2 м.д. Так как рядом находится группа СН2 сигнал проявляется в виде триплета. Протоны центральной группы СН2 дают сигнал в области 1,8 м.д. Так как на них влияют протоны и группы СН3, и группы СН2Br, мы видим секстет. Наконец, на протоны группы СН2Br влияют протоны группы СН2. Поэтому сигнал проявляется в виде триплета в области 3,4 м.д. Интегральная интенсивность Сила сигнала пропорциональна числу резонирующих протонов, вызывающих этот сигнал. Площадь пика в спектре ПМР также пропорциональна числу резонирующих протонов. В ЯМР-спектрометре имеется электронный интегратор, который дает площади пиков в виде кривой, называемой кривой интегральной интенсивности.  Рис.8. Спектр ПМР бензилового спирта с кривой интегральной интенсивности. Интегральная интенсивность используется для подтверждения правильности идентификации вещества, а также для количественного определения веществ в смесях. ИНФРАКРАСНАЯ СПЕКТРОСКОПИЯ (ИК-спектроскопия) ИК-область спектра охватывает диапазон от 4000 до 625 см-1. Соседние области называются ближней ИК-областью (от 12500 до 4000 см-1) и дальней ИК-областью (от 625 до 50 см-1). Поглощение ИК-излучения молекулой вещества вызывает изменения длин связей и углов между связями. То есть, в зависимости от частоты поглощенного излучения начинает периодически растягиваться определенная связь или искажаться определенный угол между связями. Колебания, заключающиеся в изменении длины связи между атомами, и не сопровождающиеся отклонениями атомов от межъядерной оси называются валентными.  Колебания, при которых атомы смещаются с межъядерной оси, то есть сопровождающиеся изменением углов между связями, называются деформационными.  Каждому типу связей и каждому виду колебаний соответствует определенная энергия. Значит, каждому колебанию соответствует определенная частота излучения ν. Поэтому, зная, в какой области, при какой частоте происходит поглощение излучения данным веществом, можно сделать вывод о строении вещества. Часто частоту заменяют пропорциональной ей величиной, обратной длиной волны. Единицей измерения в этом случае служит обратный сантиметр, см-1. 3.1. Принципиальная схема ИК-спектрометра.  Рис.1 Принципиальная схема ИК-спектрометра. Свет от источника излучен-1.ия с помощью двух зеркал делится на два одинаковых луча, один из которых проходит через кювету с образцом, а второй проходит через кювету с раствором сравнения. Затем каждый луч проходит через монохроматор, который позволяет выделять излучение строго определенной частоты, и попадает на детектор, где энергия излучения преобразуется в электрический ток. Обычно рассматривается область от 600 до 4000 см-1. 3.2. Расшифровка ИК-спектра. 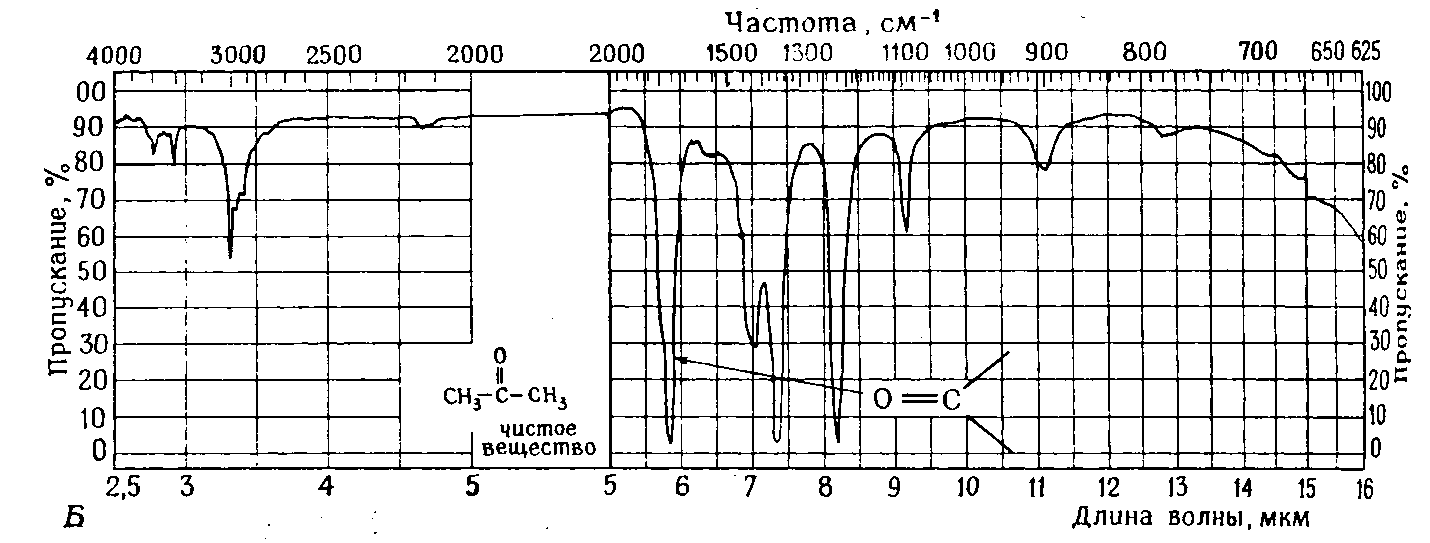 Рис.2. ИК-спектр ацетона. ИК-спектр записывается в виде кривой. На рисунке 2 приведен спектр ацетона. Расшифровка спектра заключается в поиске в спектре полос поглощения в виде пиков, характерных для данного соединения. Характеристические частоты поглощения в ИК-области
ИК-спектры очень специфичны. Они также индивидуальны для каждого соединения, как отпечатки пальцев человека. Не случайно область от 625 до 1250 см-1 называют областью отпечатков пальцев. При расшифровке ИК-спектра или его сравнения с литературным учитывается не только расположение полос поглощения в спектре, но и форма пика и его интенсивность. ИК-спектроскопия часто используется и для количественного анализа соединений. В этом случае в спектре выбирается полоса поглощения достаточной интенсивности, предварительно снимаются ИК-спектры растворов вещества определенной концентрации, строится градуировочная кривая. МАСС – СПЕКТРОСКОПИЯ При исследовании органических веществ методами ПМР- и ИК-спектроскопии молекулы вещества не подвергаются разрушению. В отличие от них масс-спектроскопия является методом, вызывающим деструкцию образца. В этом методе молекулы вещества подвергаются действию электронного удара, что приводит к их разрушению. Если молекула получает удар пучка электронов низкой энергии (около 10 эВ), она теряет электрон и превращается в молекулярный ион. Если молекула подвергается бомбардировке потоком электронов с большей энергией (около 70 эВ), то образовавшийся молекулярный ион распадается на более мелкие фрагменты, как имеющие заряд, так и незаряженные. Масс- спектроскопия исследует только заряженные фрагменты молекулы. Так как в камере масс-спектрометра создается очень низкое давление (около 10-7 мм.рт.ст.), после удара молекулы пучком электронов могут происходить лишь внутримолекулярные реакции.  4.1. Принципиальная схема масс-спектрометра. 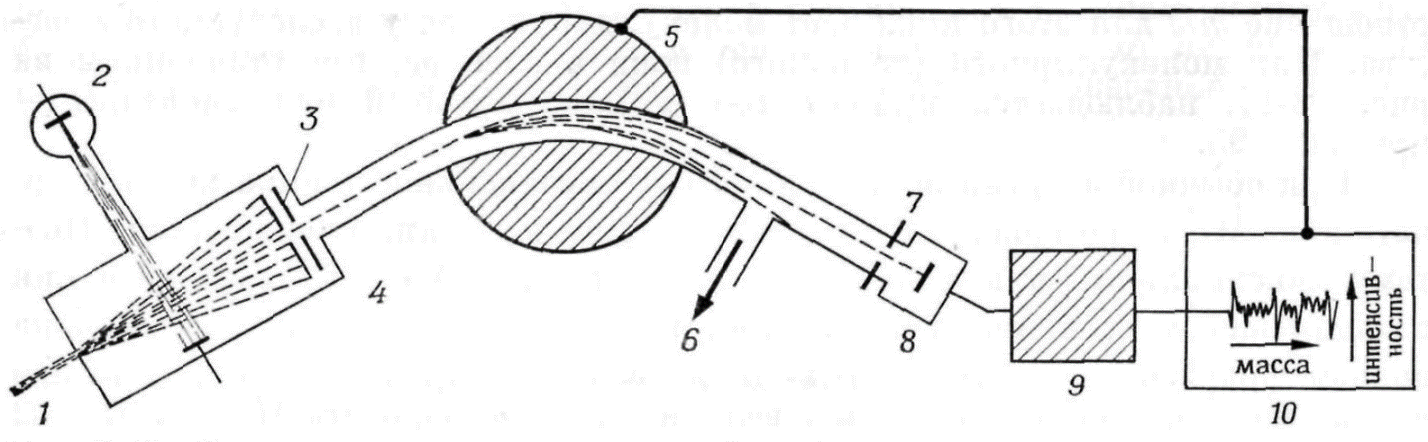 Рис.3. Принципиальная схема масс-спектрометра. 1-ввод образца; 2-источник электронов; 3-ускоряющие пластины и щели; 4-ионизационная камера; 5-магнит с переменным полем; 6-вакуумный насос; 7-щель; 8-коллектор ионов; 9- усилитель; 10-самонисец. Образец вводится в ионизационную камеру, где подвергается действию потока электронов. Затем образующиеся при этом положительные ионы разгоняются отрицательно заряженными ускоряющими пластинами 3 и попадают в анализатор. Анализирующий магнит 5 создает магнитное поле, под действием которого происходит как бы сортировка ионов в зависимости от их отношения m/e. При хорошем разрешении через щель 7 проходят и попадают на коллектор 8 только ионы одной массы. 4.2. Применение масс-спектроскопии. Масс-спектроскопия используется для определения точной массы и молекулярной формулы вещества, а также для исследования строения вещества. При определении молекулярной массы образец бомбардируют электронами с низкой энергией. В результате получаются молекулярные ионы, которые затем идентифицируются. Масс-спектр изображает зависимость интенсивности сигнала от отношения m/e для различных заряженных продуктов, образующихся при распаде вещества. Так как заряд электрона равен 1, отношение m/e обычно сводится к массе, то есть массе иона. На рисунке 4 приведен масс-спектр α-пинена, который облучался потоком электронов с энергией 70 эВ.  Рис.4. Масс спектр α-пинена. Пик молекулярного иона m/e 136 менее интенсивный, что объясняется тем, что молекулярный ион при облучении энергией 70 эВ претерпевает дальнейшие превращения и его интенсивность в спектре уменьшается. Чтобы проиллюстрировать характер превращений молекулярного иона рассмотрим масс-спектр гексана, приведенный на рисунке5.  Рис.5. Масс-спетр гексана. Общий вид спектра характеризуется распределением пиков ионов группами с интервалом в 14 единиц массы., что отвечает отрыву метиленовой группы СН2. Такой вид масс-спектра типичен для неразветвленных насыщенных углеводородов. Максимальный пик в каждой группе отвечает катиону, образующемуся при потере алкильного радикала. Менее интенсивные пики возникают при потере алкильного радикала. Еще менее интенсивные пики возникают при дальнейшей потере атомов водорода. Можно предположить следующую схему распада:  Очень часто масс-спектроскопия применяется в органической химии в сочетании с газовой хроматографией. Результаты обрабатываются с помощью компьютера. Этот метод является в настоящее время наиболее мощным методом разделения и анализа химических соединений. |