Электронные эффекты. Лекция 5 Схема лекции
 Скачать 0.6 Mb. Скачать 0.6 Mb.
|

|
ЛЕКЦИЯ 5 Схема лекции. 5.1. Электронные эффекты в органической химии. 5.1.1. Индуктивный эффект. 5.1.2. Эффекты сопряжения. 5.1.3. Резонанс. 5.2. Теория кислот и оснований в органической химии. 5.2.1. Кислоты Бренстеда. 5.2.2. Основания Бренстеда. 5.2.3. Кислоты и основания Льюиса. 5.1. ЭЛЕКТРОННЫЕ ЭФФЕКТЫ В ОРГАНИЧЕСКОЙ ХИМИИ. Химические свойства органических веществ различных рядов и классов определяются особенностями распределения электронной плотности в молекулах этих веществ, что в свою очередь определяется строением молекул и наличием функциональных групп. Строение функциональных групп и природа атомов, входящих в их состав находят свое отражение в так называемых электронных эффектах. К электронным эффектам относятся: индуктивный эффект, эффект поля (передача воздействия через пространство электростатическим путем), эффект сопряжения и эффект отталкивания орбиталей. Мы рассмотрим индуктивный эффект и эффект сопряжения. 5.1.1. Индуктивный эффект. Среди свойств ковалентной связи отмечалось свойство полярности, что выражается в смещении пары электронов, образующих связь в строну более электроотрицательного атома. Мерой полярности ковалентной связи является дипольный момент. Дипольный момент связи оказывает воздействие на соседнюю связь. Эта связь в свою очередь оказывает воздействие на соседнюю связь и т.д. В итоге происходит смещение электронных облаков химических связей вдоль линии σ-связей, вызванное различием в значениях электроотрицательности атомов. Это явление смещения получило название индуктивного эффекта. Определение: Индуктивным эффектом называется смещение электронной плотности σ-связей вдоль линии связи, вызванное различием в значениях электроотрицательности атомов. Индуктивный эффект имеет электростатическую природу. Он передается по линии связи и ведет к появлению дробных электрических зарядов на атомах. Заряды обозначаются символами δ+ и δ-. Степень смещения электронной плотности зависит от разности электроотрицательности соответствующих атомов. Знак индуктивного эффекта определяется относительно атома или группы атомов, принятого за эталон. В качественных оценках его часто определяют относительно атома водорода. При количественном рассмотрении за стандарт принята группа СН3-. Индуктивный эффект изображают стрелкой вдоль σ-связи. Стрелка указывает направление смещения электронной плотности. Индуктивный эффект передается по цепи σ-связей с затуханием. Наиболее подвержен действию индуктивного эффекта α-углеродный атом. Заместители, притягивающие электроны, характеризуют отрицательным индуктивным эффектом. Их называют электроноакцепторными заместителями. Примерами электроноакцепторных заместителей, обладающих отрицательным индуктивным эффектом (-I-эффект). являются: F-; Cl-; Br-; -OH; -ОR; -COOH; -CHO; -COOR; -CN; -NO2; -NH2; СН2=СН-; С6Н5-; СНС- В состав данных функциональных групп входят атомы с электроотрицательность более высокой, чем электроотрицательность атома углерода в метильном радикале.  Заместители, отталкивающие электроны от себя проявляют положительный индуктивный эффект (+I-эффект). Такие заместители называются электронодонорными. Положительный индуктивный эффект также определяется относительно метильной группы. Положительным индуктивным эффектом обладают атомы металлов, а также разветвленные алкильные группы:  Заместители, в которых находится атом углерода в состояниях sp2-гибридизации и sp-гибридизации являются электроакцепторами по сравнению с атомом углерода в состоянии sp3-гибридизации и, соответственно, обладают отрицательным индуктивным эффектом. Атомы с целым отрицательным зарядом обладают положительным индуктивным эффектом: Атомы с целым положительным зарядом обладают отрицательным индуктивным эффектом. 5.1.2. Эффекты сопряжения. Осуществление влияния природы атомов на распределение электронной плотности в молекуле возможно как по системе σ-связей посредством индуктивного эффекта, так и по системе π-связей. Способность π-электронов к смещению особенно ярко выражена в соединениях, в которых существует система чередующихся кратных и простых связей: 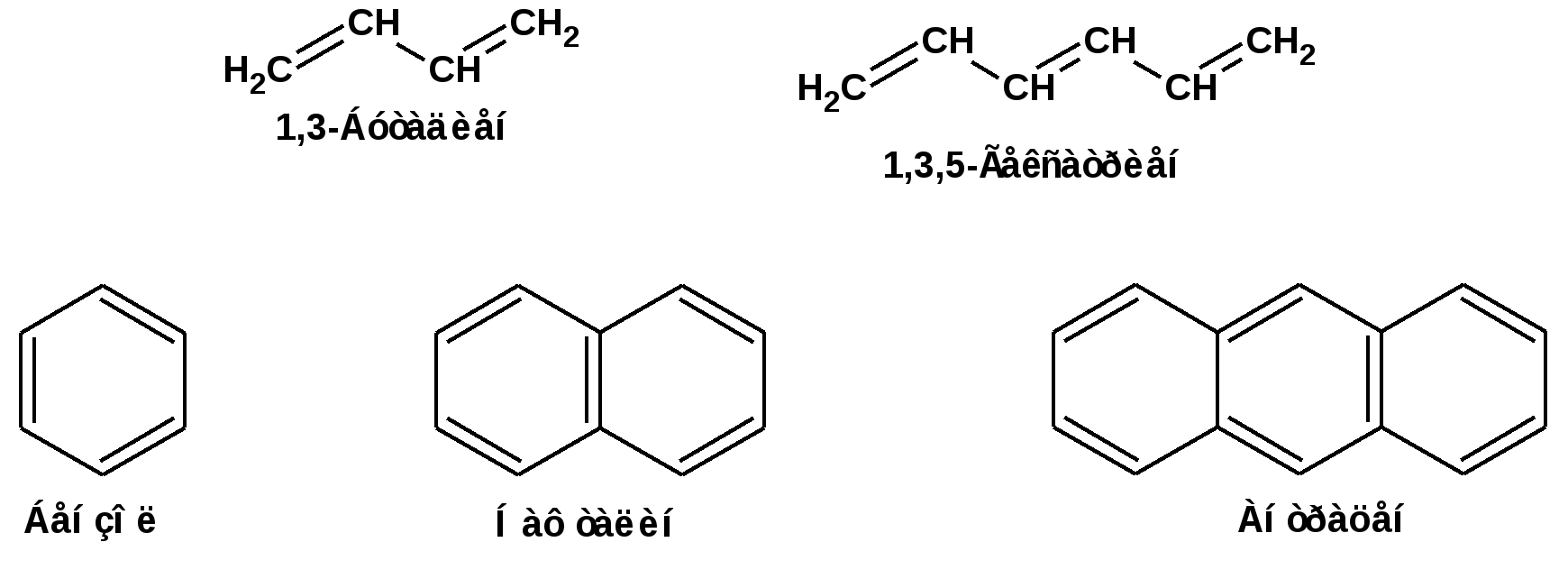 Такие соединения построены только из атомов в состоянии sp2-гибридизации. А сами такие системы называются сопряженными. Физической основой сопряжения является взаимодействие (перекрывание) р-орбиталей соседних двойных связей: 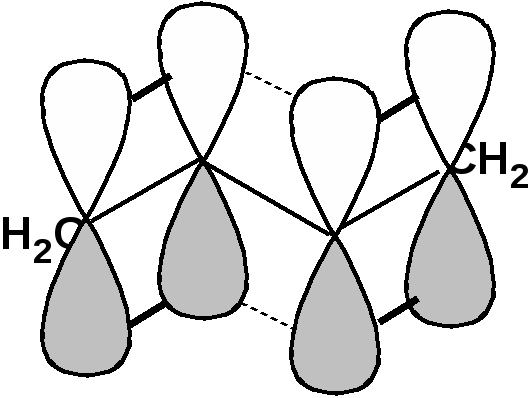 Сопряженная система представляет собой сплошную π-связь только электронная плотность концентрированна в большей степени на двойной связи и в меньшей степени на простой связи. Т.е. в случае сопряженной системы отсутствуют разрывы в цепочке связей как в случае σ-остова. Соответственно в случае появления в сопряженной системе атома, электроотрицательность которого отличается от электроотрицательности атома углерода в состоянии sp2-гибридизации, то в зависимости от того донором или акцептором является функциональная группа – произойдет смещение электронной плотности по всей цепи сопряжения. Эффект смещения электронной плотности по цепи сопряжения называется мезомерным эффектом (обозначается буквой «М»). В зависимости от направления смещения электронной плотности от заместителя или к нему различают положительный мезомерный эффект (+М) и отрицательный мезомерный эффект (-М). Графически мезомерные эффекты обозначается изогнутой стрелкой Часто термин «мезомерный эффект» заменяют термином «эффект сопряжения». Но сопряжение обозначает перекрывание орбиталей соседних связей, а мезомерия обозначает передачу влияния по цепи сопряженных связей: 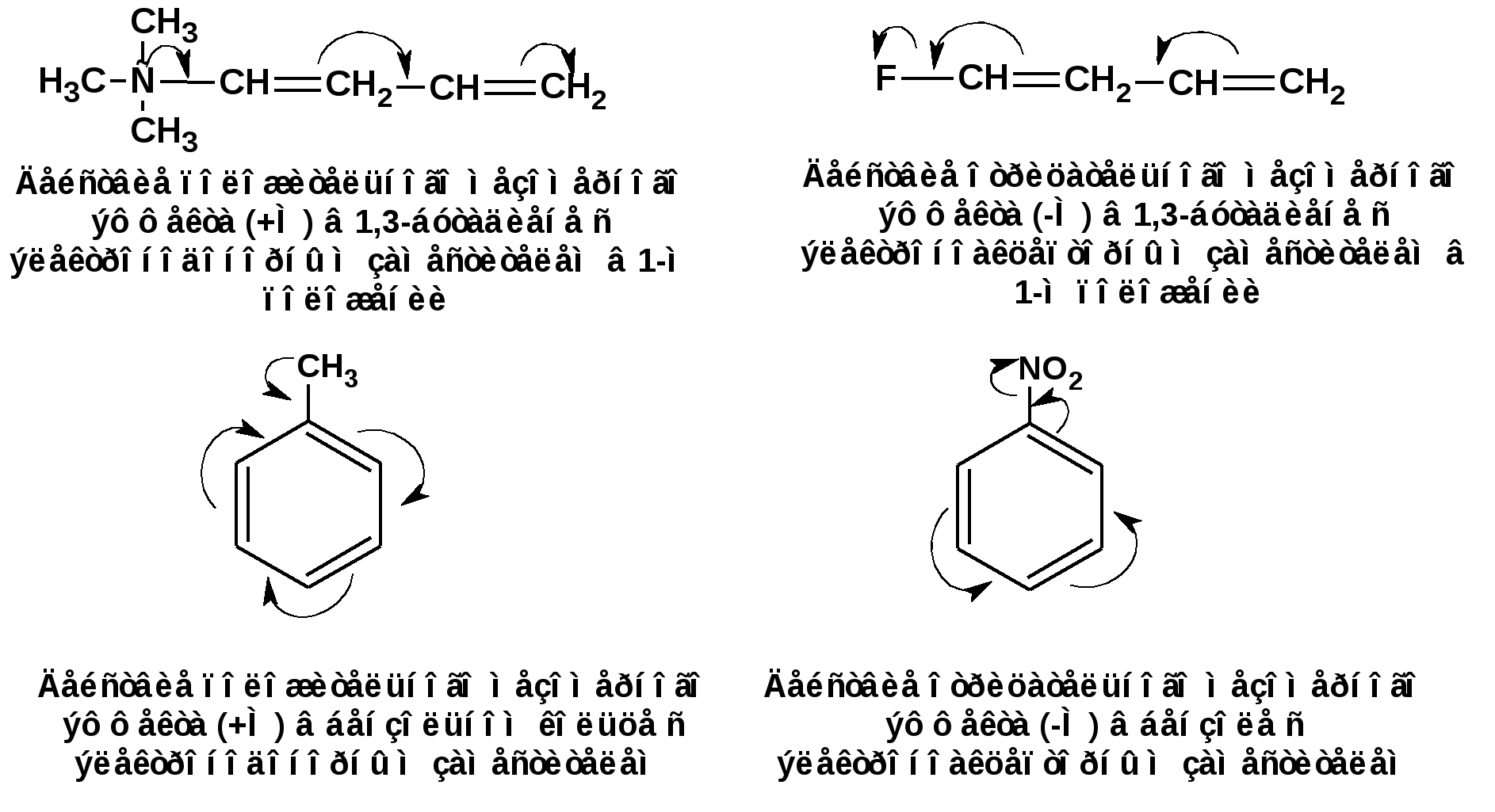 Комбинация изогнутых стрелок по структурной формуле отражает перераспределение электронной плотности в цепи сопряжения. Данное перераспределение называется делокализацией «размазыванием». Иногда делокализацию изображают графически пунктирной линией по структурной формуле: 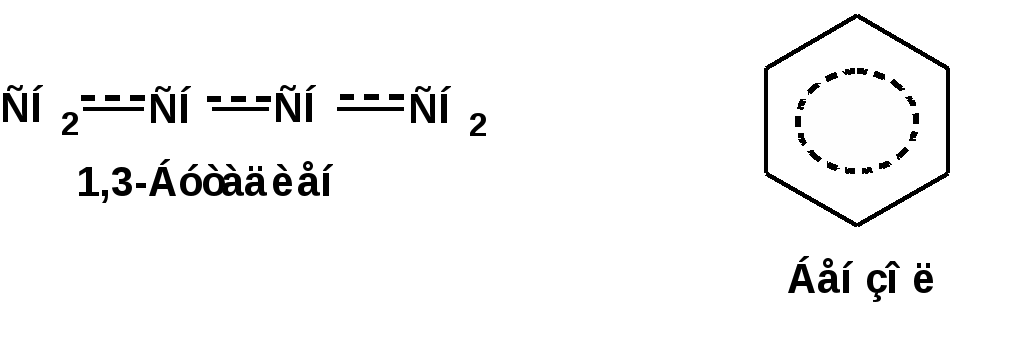 Такие формулы называют мезомерными формулами. Делокализация оказывает большое влияние на свойства сопряженных молекул. Чем выше степень делокализации тем выше термодинамическая стабильность сопряженной системы. Обратите внимание на порядок в своих комнатах. Чаще бывает беспорядок. Т.е. беспорядок более устойчивое состояние. Делокализация - то беспорядок в распределении электронов. Частным случаем делокализации является сверхсопряжение, которое способствует стабилизации неспаренных электронов алкильных радикалов. Эффекты сопряжения характерны для соединений с кратными связями (двойные, тройные) независимо от природы атомов, соединенных кратными связями, а также для функциональных групп в состав которых входят гетероатомы с неподеленными электронными парами. 5.1.3. Резонанс. Удобным способом изображения делокализации в сопряженных системах является изображение с помощью резонансных структур . При написании резонансных структур следует соблюдать следующие правила: 1. Атомы и молекулы не меняют своего положения; изменяется положение НЭП и π-электронов кратных связей. 2. Каждая резонансная структура, приписываемая данному соединению, должна иметь одну и ту же сумму π-электронов, включая π-связи и НЭП. 3. Между резонансными структурами ставят резонансную стрелку «↔». 4. В резонансных структурах не принято обозначение электронных эффектов при помощи прямых и изогнутых стрелок. 5. Набор резонансных структур молекулы, иона или радикала следует заключать в квадратные скобки. Например: 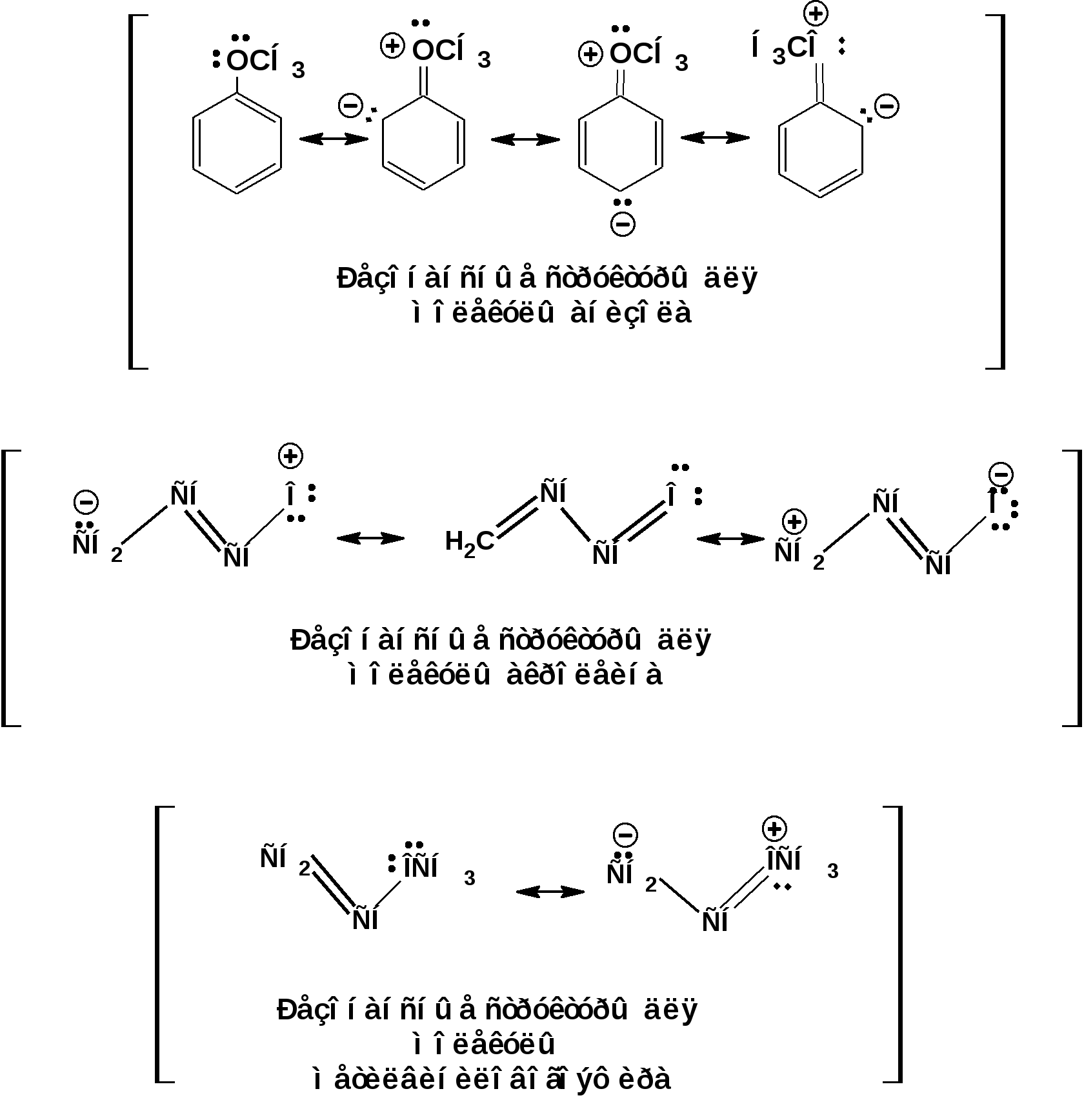 При оценке резонансной стабилизации молекул и частиц, а также при сравнении относительных энергий различных резонансных структур необходимо руководствоваться следующими правилами: 1. Энергия реальной молекулы меньше. Чем энергия любой из резонансных структур. 2. чем больше резонансных структур можно написать для данной молекулы или частицы, тем она стабильнее. 3. При прочих равных условиях более стабильными являются резонансные структуры с отрицательным зарядом на наиболее электроотрицательном атоме и с положительным зарядом на наиболее электроположительном атоме. 4. Резонансные структуры, в которых все атомы имеют октет электронов, более стабильны. 5. максимальную стабильность имеют частицы, для которых резонансные структуры являются эквивалентными, а соответственно имеют одинаковую энергию. 5.2. ТЕОРИЯ КИСЛОТ И ОСНОВАНИЙ В ОРГАНИЧЕСКОЙ ХИМИИ В органической химии действуют две основные теории кислот и оснований. Это теории Бренстеда и Льюиса.Определение: Согласно теории Бренстеда кислотой является любое вещество, способное диссоциировать с отщеплением протона. Т.е. кислота – это донор протонов. Основанием является любое вещество, способное присоединять протон. Т.е. основание – это акцептор протонов. Согласно теории Льюиса кислотой является любая молекула или частица, способная принимать электроны на вакантную орбиталь. Т.е. кислота – это акцептор электронов. Основанием является любая молекула или частица, способная быть донором электронов. Т.е. основание – это донор электронов.Определение: Частица, образующаяся из кислоты после диссоциации и несущая отрицательный заряд - называется сопряженным основанием. Частица, образующаяся из основания после присоединения протона и несущая положительный заряд - называется сопряженной кислотой. 5.2.1. Кислоты Бренстеда Характеристикой силы кислот, по отношению к воде, является константа диссоциации, являющаяся константой равновесия следующей реакции: 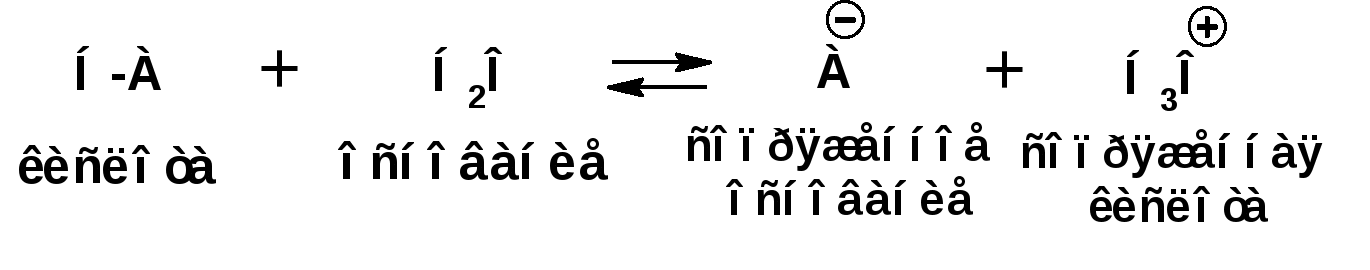 Наиболее известные примеры кислот в органической химии это карбоновые кислоты алифатические, например уксусная кислота: 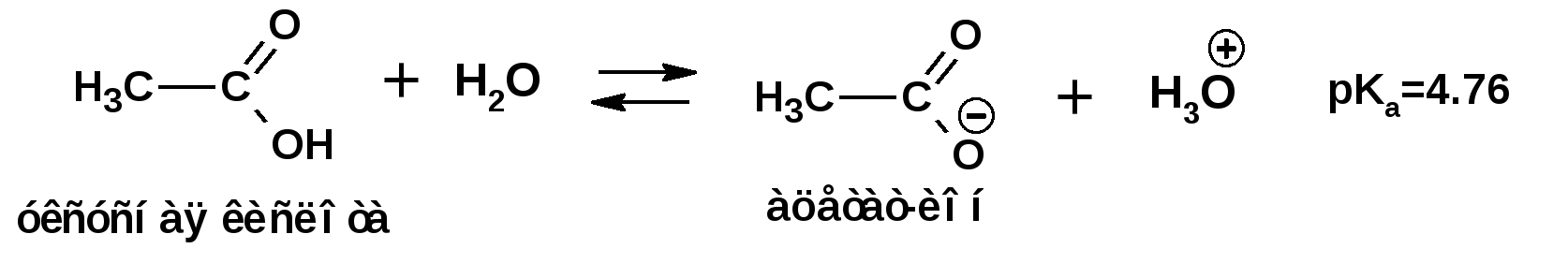 и бензойная: 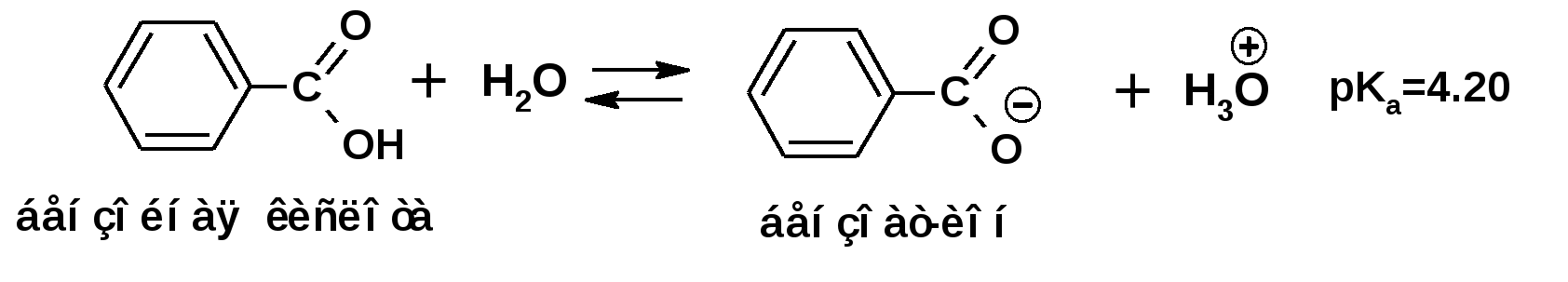 Карбоновые кислоты являются кислотами средней силы. В этом можно убедиться сравнивая значения рК карбоновых кислот и некоторых других приведенных ниже: 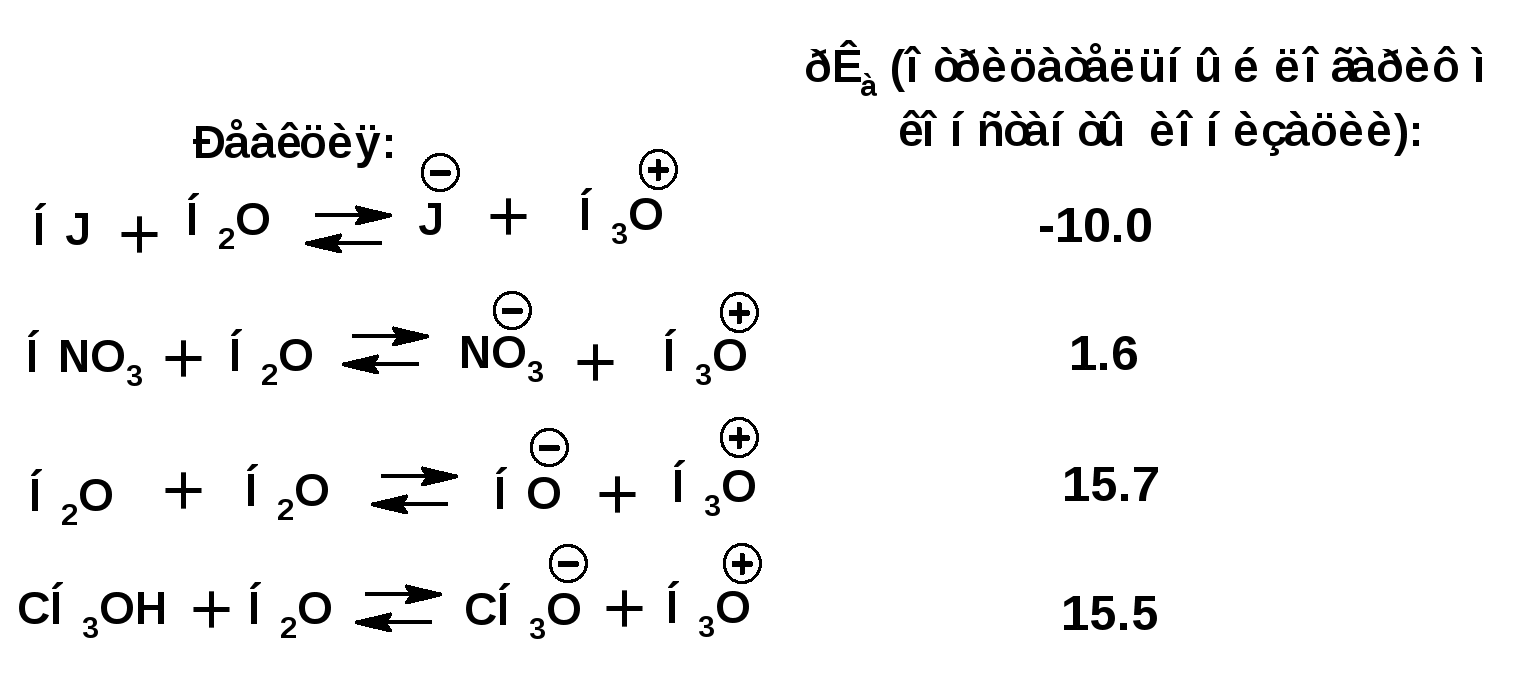 Отщеплять протон могут органические соединения, относящиеся к разным классам органических соединений. Среди органических соединений различают ОН-, SH-, NH- и СН-кислоты. К ОН-кислотам относятся карбоновые кислоты, спирты и фенолы. К NH-кислотам относятся амины и амиды. К СН-кислотам относятся нитроалканы, карбонильные соединения, сложные эфиры, терминальные алкины. В очень слабым СН-кислотам относятся алкены, ароматические углеводороды и алканы. Сила кислоты тесно связана с устойчивостью сопряженного основания. Чем устойчивее сопряженное основание, тем более кислотно-основное равновесие смещено в строну сопряженных основания и кислоты. Стабилизация сопряженной кислоты может быть обусловлена следующими факторами: Чем выше электроотрицательность атома, тем сильнее он удерживает он электроны в сопряженном основании. Например, рК фтористого водорода 3.17; рК воды 15.7; рК аммиака 33 и рК метана 48. 2. Стабилизация аниона по мезомерному механизму. Например, в карбоксилат-анионе: 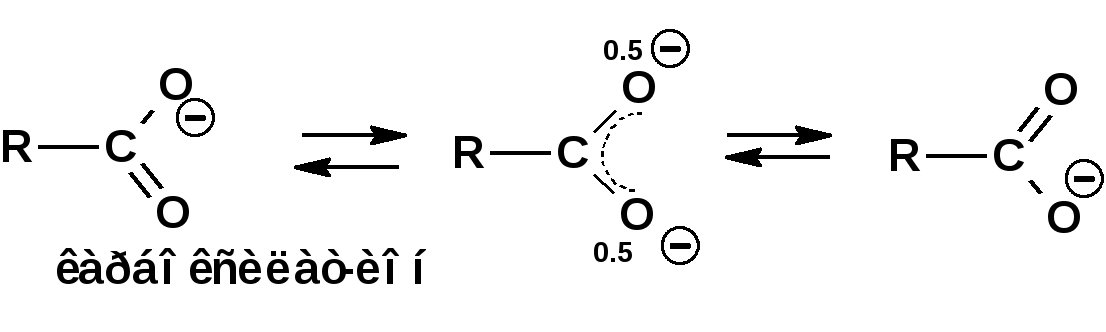 В алкоксид-ионе, например: 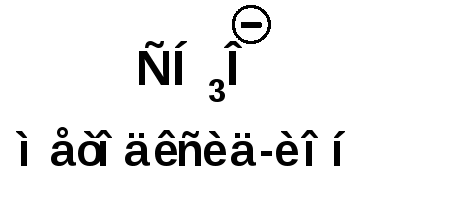 такая стабилизация невозможна. Соответственно для уксусной кислоты рК=4.76, рК метилового спирта 15.5. Другим примером стабилизации сопряженного основания является фенолят-ион, образующийся в результате диссоциации фенола:  Для образовавшегося феноксид (или фенолят)-иона можно построить резонансные структуры, отражающие делокализацию отрицательного заряда по ароматическому кольцу: 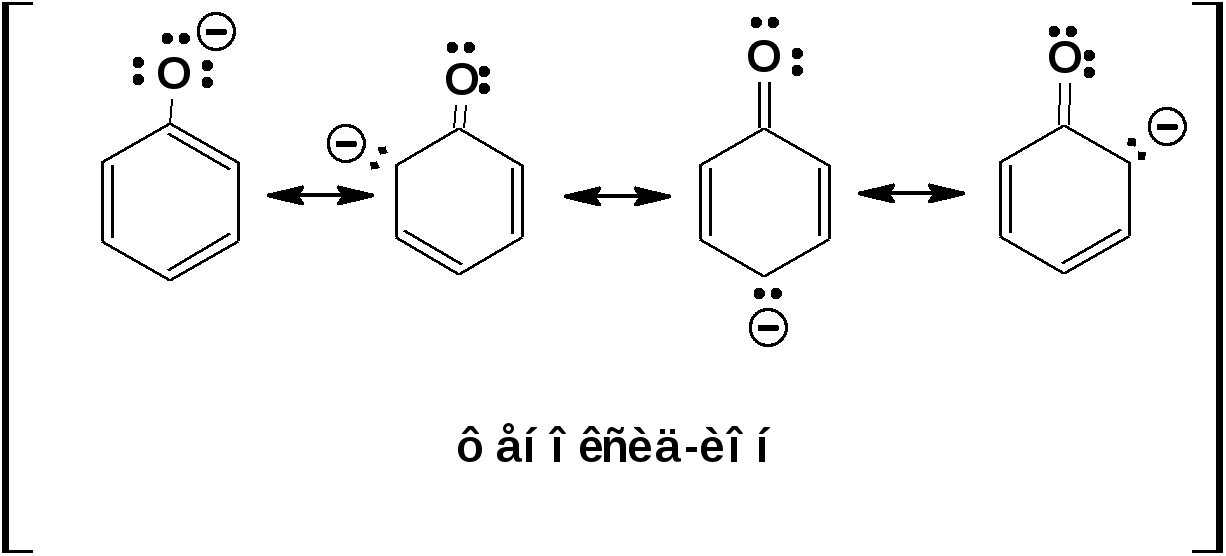 Соответственно рК фенола равно 9.98, а метанола, для которого невозможно построить резонансные структуры имеет рК равное 15.5. 3. Введение электронодонорных заместителей дестабилизирует сопряженное основание и соответственно снижает силу кислоты: 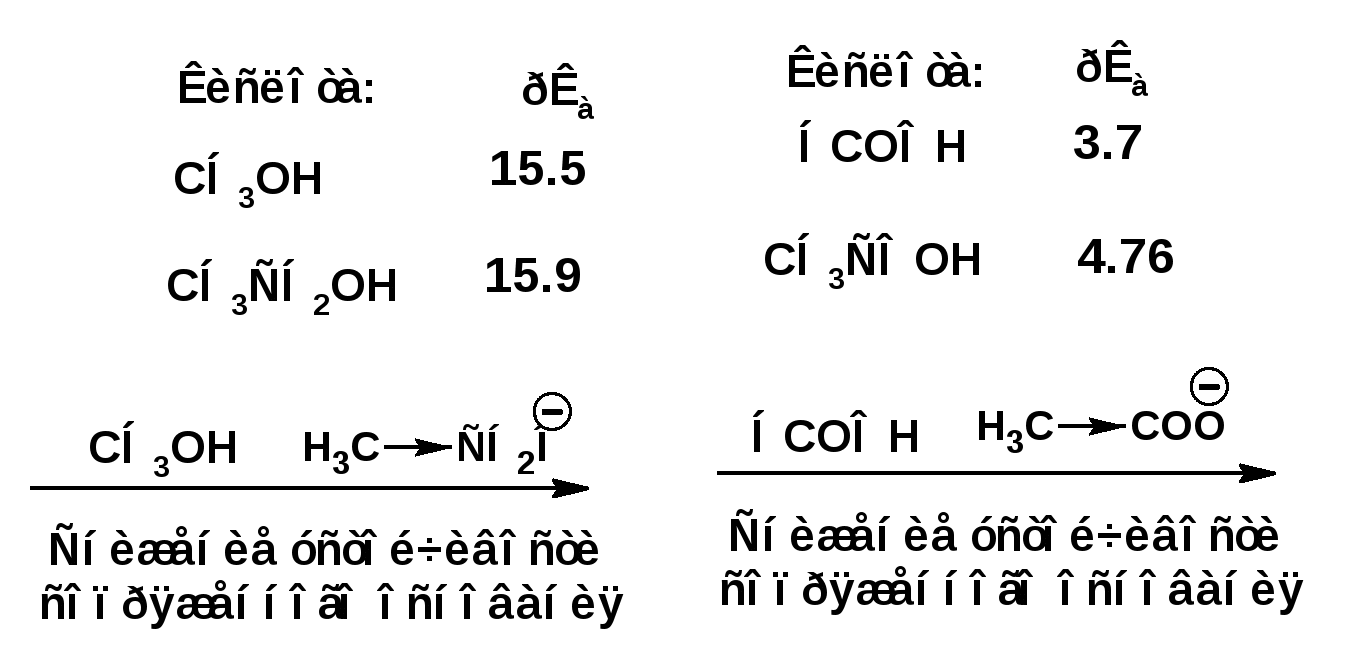 4. Введение электроноакцепторных заместителей стабилизирует сопряженное основание и повышает силу кислот:  5. Удаление по цепи электроноакцепторного заместителя от протонодонорной группы ведет к снижению силы кислоты:  Приведенные данные иллюстрируют быстрое затухание индуктивного эффекта с увеличением углеводородной цепи. Особое внимание следует уделить СН-кислотам, поскольку, образующиеся при их диссоциации сопряженные основания, в качестве которых выступают карбанионы. Эти нуклеофильные частицы являются промежуточными во многих органических реакциях. СН-кислоты наиболее слабые из кислот всех типов. Продуктом кислотной диссоциации является карбанион – частица, в которой основой является атом углерода , несущий отрицательный заряд. Такая частица имеет тетраэдрическое строение. НЭП занимает sp3-гибридную орбиталь. Сила СН-кислоты определяется теми же факторами, чито и сила ОН-кислоты. Ряд стабилизирующего влияния заместителей совпадает с рядом увеличения их электроноакцепторных свойств: 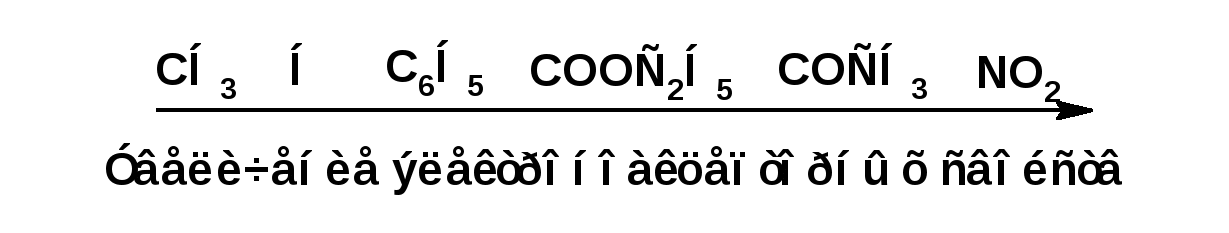 Среди СН-кислот особый интерес представляют аллил-анион и бензил-анион. Эти анионы можно представить в форме резонансных структур: 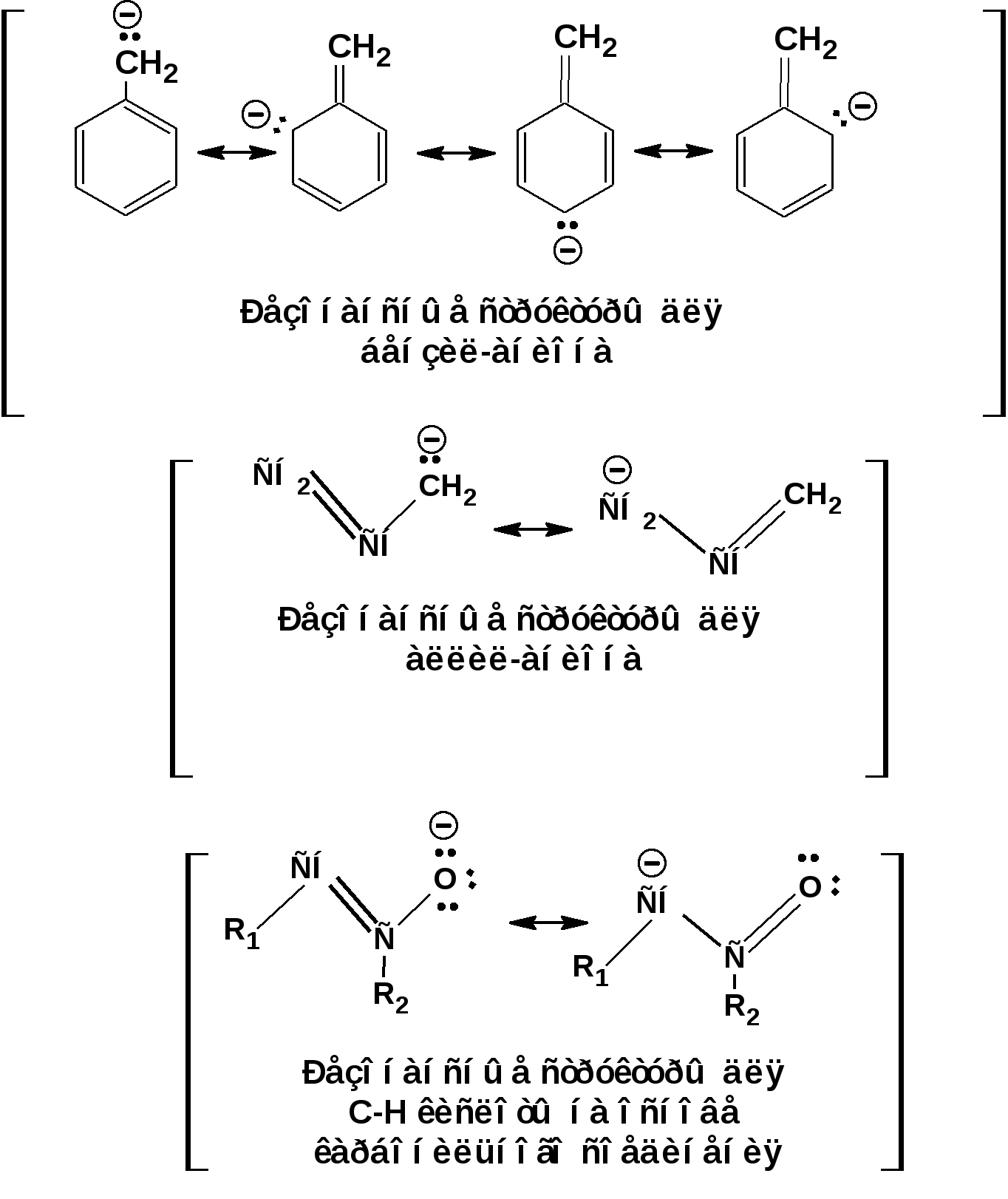 Эффект делокализации отрицательного заряда в бензил-анионе столь велик, что его геометрия приближается к плоской. При этом углеродный атом карбанионного центра меняет гибридизацию с sp3 на sp2. 5.2.2. Основания Бренстеда. Для оснований Бренстеда действуют те же закономерности, что и для кислот. Также рассматривается кислотно-основная реакция, которая приводит к образованию сопряженной кислоты:  Константу равновесия обозначают как Кb Чем больше Кb , тем выше сила основания. Более универсальной оценкой силы органического основания, является рКа его сопряженной кислоты - рКа(ВН+): 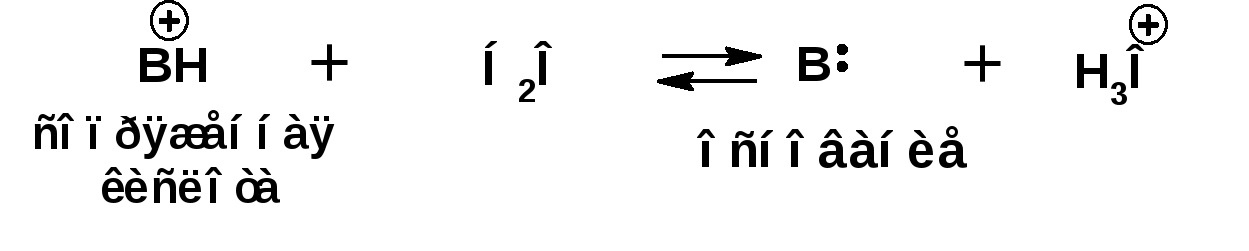 Соответствующая константа и рК: 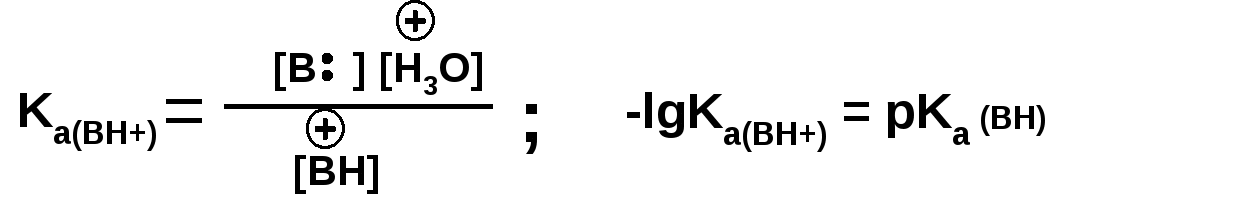 Слабым основаниям соответствуют сильные сопряженные кислоты и наоборот: сильным основаниям соответствуют слабые сопряженные кислоты. Типичными органическими основаниями по Бренстеду являются амины. При этом алифатические амины значительно более сильные основания, чем ароматические. 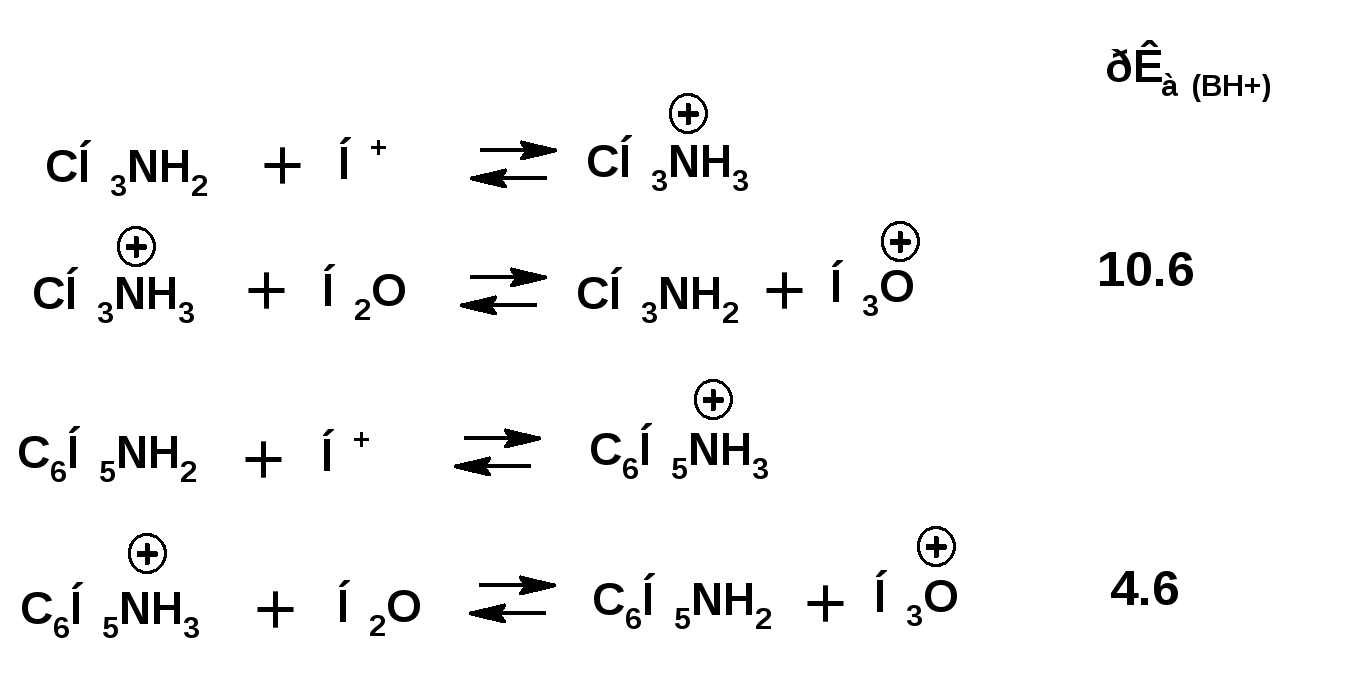 В молекуле анилина НЭП атома азота сопряжена с π-электронами бензольного кольца. Этот эффект повышает силу его сопряженной кислоты и соответственно снижает основность анилина по сравнению с метиламином: 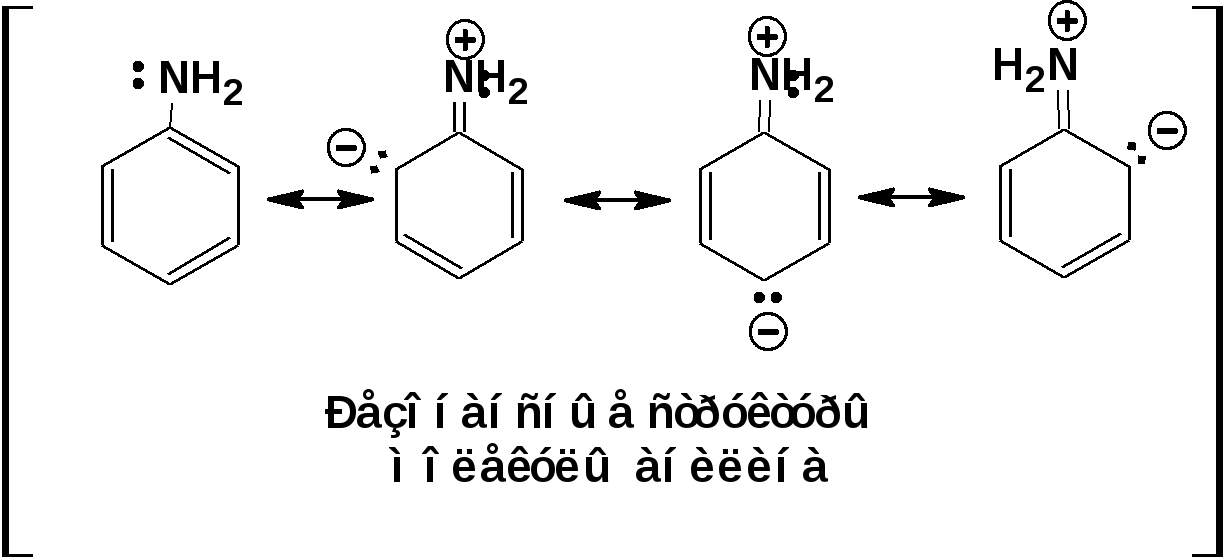 Для сопряженной кислоты можно нарисовать резонансные структуры, следовательно сопряженная кислота устойчивая, а соответствующее основание слабое. 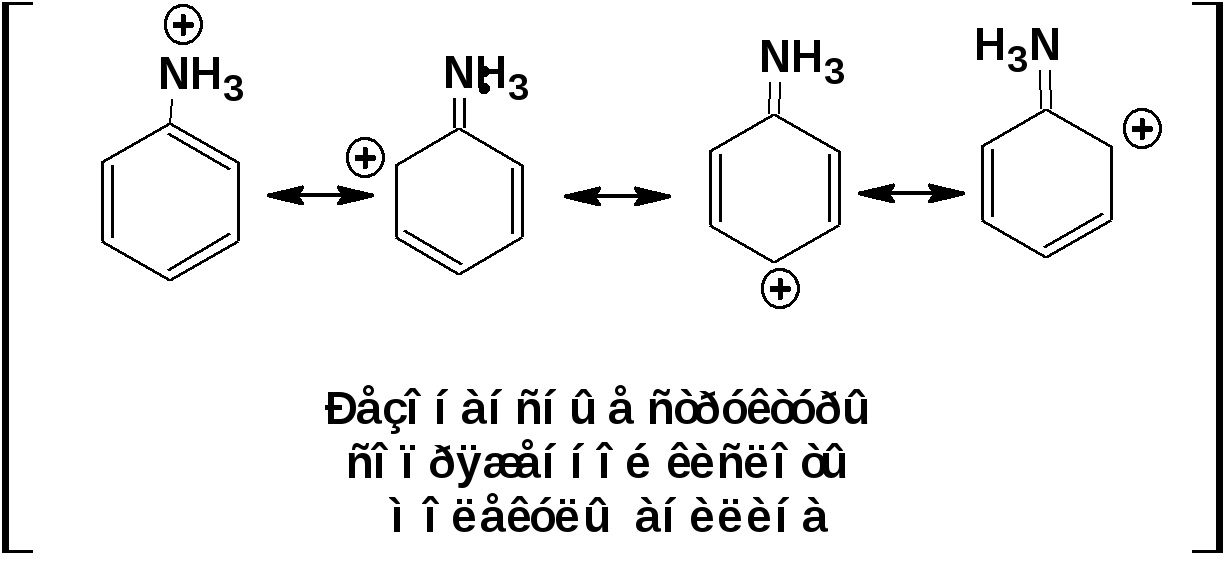 В молекуле метиламина НЭП полностью локализована на атоме азота. Это способствует снижению силы его сопряженной кислоты, а, следовательно, повышает основность метиламина. Кислоты Льюиса: Кислота Льюиса – это любая молекула или частица, способная принимать (акцептировать) электроны на вакантную орбиталь. В качестве вакантной орбитали выступает, как правило, низшая свободная молекулярная орбиталь (НСМО). НСМО может быть π- и σ-орбиталь. Соответственно среди кислот Льюиса различают π- σ-кислоты. Кислотами Льюиса могут быть частицы и молекулы не несущие электрического заряда: BF3; AlCl3; AlBr3;FeCl3; ZnCl2; SnCl4; TiCl3; TiCl4.. Частицы несущие положительный заряд (катионы): σ-Кислотой является протон. Большинство кислот Льюиса являются π-кислотами. К π-кислотам относятся алкены и арены имеющие электроноакцепторные заместители. Например:  5.2.4. Основания Льюиса. Основаниями Льюиса являются любые частицы, способные выступать в роли доноров пары электронов. Как правило, донорной орбиталью явояется высшая занятая молекулярная орбиталь (ВЗМО). В зависимости от характера ВЗМО различают: 1. π-доноры (π-основания) 2. n-доноры (n -основания) 3. σ- доноры (σ -основания) В π-донорах высшей занятой молекулярной орбиталью является π-орбиталь. Примерами π-доноров являются алкены, сопряженные алкадиены, бензол и его производные, другие арены. В n-донорах донорной орбиталью является орбиталь, заселенная НЭП. Примерами n -доноров являются спирты, амины, алкоксид-анионы, амид-анионы. В σ-донорах высшей занятой молекулярной орбиталью является σ –орбиталь. Свойства σ -доноров проявляют алканы в реакциях с участием сверхкислот и комплексов переходных металлов. |