Антибиотики. Основные механизмы устойчивости к антибиотикам у бактерий
 Скачать 188 Kb. Скачать 188 Kb.
|

|
Основные механизмы устойчивости к антибиотикам у бактерий М.А. Петрова (ИМГ РАН) Устойчивость бактерий к антибиотикам может быть врожденной (intrinsic) и приобретенной. Врожденная (природная) устойчивость характеризуется отсутствием у микроорганизмов мишени действия антибиотика или недоступностью мишени вследствие исходно низкой проницаемости или ферментативной инактивации. Приобретенная (вторичная) устойчивость возникает в результате контакта микроорганизма с антимикробным средством либо за счет возникновения мутаций хромосомной ДНК, модифицирующих имеющиеся белки бактерий, либо за счет трансформации, благодаря которой образуются мозаичные гены, либо путем горизонтального переноса генов (ГПГ) устойчивости. Разные авторы по-разному определяют по существу одни и те же механизмы устойчивости. Наиболее хорошо изучены и распространены 4 основных биохимических механизма устойчивости бактерий к антибиотикам: (1) энзиматическая инактивация антибиотика; (2) модификация молекулы-мишени действия антибиотика; (3) активное выведение антибиотика из микробной клетки и (4) изменение проницаемости внешней мембраны микробной клетки. Последние два механизма некоторые исследователи объединяют в один, считая, что оба из них обуславливают ограничение доступа антибиотика к мишени, тогда как первый из перечисленных механизмов напротив иногда подразделяют на модификацию антибиотика и его деградацию. Кроме этих основных типов в последние годы обнаружены еще и другие механизмы устойчивости, например формирование метаболического «шунта» (приобретение генов метаболического пути альтернативного тому, который ингибируется антибиотиком), имитация молекулы-мишени, сверхэкспрессия молекулы-мишени (Hegde et al., 2005; Nikaido., 2009; van Hoek et al., 2011). 4.2.1. Энзиматическая инактивацияНаиболее распространены реакции энзиматической инактивации молекул антибиотика. Будучи очень разнообразными подобные механизмы функционируют в отношении антибиотиков, относящихся к самым различным классам химических соединений, таких как ß-лактамы, аминогликозиды, хлорамфеникол, эритромицин, линкомицин и родственные им соединения (табл. 4.1.). Инактивация происходит за счет синтеза ферментов, способных специфично реагировать с антибиотиком и модифицировать его, либо нарушая его аффинность к мишени, либо необратимо связываясь и не позволяя прореагировать с мишенью, либо полностью инактивируя или разрушая молекулу антибиотика (Wright, 2005; Queenan et al., 2010; Ramirez et al., 2010; Singh et al., 2012). Механизмы_энзиматической_инактивации_антибиотиков_(по_Wright,_2005)'>Таблица 4.1. Механизмы энзиматической инактивации антибиотиков (по Wright, 2005)
4.2.2. Модификация молекулы-мишениДругой, не менее распространенный тип устойчивости связан с изменением структуры молекулы-мишени, с которой связывается антибиотик. Данный тип устойчивости может быть обусловлен двумя разными механизмами. Первый из них связан с возникновением спонтанных генных мутаций, приводящих к структурным изменениям кодируемых им молекул-мишеней, нарушающими связывание с антибиотиком, и стабилизацией таких мутаций в присутствии антибиотиков. Наиболее яркими примерами являются мутации в генах, кодирующих, рибосомальный белок RpsL, β-субъединицу ДНК-зависимой РНК-полимеразы и фермент ДНК-гиразу, что придаёт клетке устойчивость к стрептомицину, рифамицину и хинолонам, соответственно. При таком механизме устойчивость не может передаваться путем ГПГ (Hooper, 2000; Nikaido, 2009). Второй способ устойчивости по данному механизму обусловлен наличием генов, которые могут передаваться с помощью горизонтального переноса. Продукты этих генов модифицируют молекулу-мишень. В этом случае в результате модификации мишени процесс связывания с ней антибиотика либо частично, либо полностью нарушается. Подобные механизмы описаны в отношении эритромицина и линкомицина, т.е. антибиотиков, нарушающих функционирование рибосом. Например, метилирование рибосомной РНК эффективно защищает бактериальную клетку от летального действия эритромицина. (Robertset al., 1999; Poehlsgaard et al., 2005; 2006; Kohanski et al., 2010). Еще одним способом устойчивости по данному механизму является приобретение генов менее чувствительной молекулы-мишени от других видов. Подобный механизм устойчивости к пенициллину распространен среди устойчивых штаммов Streptococcus pneumoniae и Neisseria meningitidis, содержащих мозаичные гены DD-транспептидазы, являющейся мишенью для пенициллина (Spratt, 1994). 4.2.3. Ограничение доступа антибиотика к мишениТакже широко встречается устойчивость, обусловленная снижением содержания антибиотика в клетке, и, следовательно, доступа его к мишени. Этот механизм может осуществляться двумя основными способами: благодаря (1) активному выведению антибиотика из микробной клетки и (2) нарушению проницаемости внешних мембран микробной клетки (Nikaido, 2009). Важно отметить, что большинство авторов, описывающих устойчивость к антибиотикам за счет изменения в проницаемости внешней мембраны, всегда подчёркивают ассоциированность данного механизма с повышенным уровнем выброса молекул антибиотика из клетки, и что эти два способа повышения резистентости действуют сопряжённо и ко-регулируют друг друга. (Nikaido, 2003; Nikaido, 2009; Fernández et al., 2011). Механизм устойчивости, обусловленный активным выведением молекул антибиотика из клетки (active efflux from cell), основывается на работе специализированного набора белков, образующих так называемые трансмембранные помпы. Такие трансмембранные помпы способны транспортировать токсичные вещества, ксенобиотики, в том числе и антибиотики большинства известных на данный момент классов, за исключением гликопептидов, из внутриклеточного пространства во внешнюю среду (Marquez, 2005). Исследования в этой области получили основательный толчок лишь в последнее десятилетие благодаря полной расшифровке геномов многих, и в первую очередь патогенных, микроорганизмов. На данный момент описано 5 основных семейств бактериальных протонных помп (Marquez, 2005; Biswas et al., 2008; Li et al., 2010): ABC суперсемейство АТФ-связывающих кассет ( ATP-binding Cassette); MFсуперсемейство(Major Facilitator Superfamily); MATE семейство(Multidrug And Toxic Compound Extrusion); SMR семейство(Small Multidrug Resistance); RND суперсемейство(Resistance-Nodulation-Division); Подавляющее большинство транспортёров, ответственных за устойчивость к широкому спектру антибиотиков, принадлежат к MF и ABC суперсемействам (Zgurskaya, 2002). Последнее характеризуется высокой специфичностью по отношению к антибиотикам и часто обнаруживается у продуцентов антибактериальных агентов и некоторых других грамположительных бактерий, таких как стафилококки и энтерококки, обеспечивая их устойчивость к макролидам и другим схожим по строению веществам. MF суперсемейство менее специфично и является основным типом транспортёров, ответственных за множественную устойчивость у грамположительных бактерий. Третье по величине суперсемейство RND включает в себя большинство типов трансмембранных помп, характерных для клинических штаммов грамотрицательных бактерий (Zgurskaya, 2002; Li et al., 2010). Изменение свойств мембраны за счет изменения ее химического состава, приводящего к снижению проницаемости мембраны для антибиотиков и других химических соединений, также является одним из механизмов устойчивости у грамотрицательных бактерий (Nikaido, 2001). Внешняя мембрана (ВМ) играет огромную роль, представляя дополнительный физический барьер для защиты, не изменяя при этом эффективность обмена веществ с внешней средой. ВМ представляет собой сложную комбинацию макромолекулярных соединений, в связи с чем полная её структура была установлена лишь в последние годы. Сочетая чрезвычайно гидрофобный липидный бислой с поринами (белками, образующими поры), имеющими специфические параметры проницаемости, ВМ действует как селективный барьер. Таким образом, проницаемость такого барьера сильно влияет и на чувствительность бактериальной клетки к антибиотикам. Небольшие по размеру гидрофильные органические вещества, как например β-лактамы, попадают внутрь клетки через порины, тогда как макролиды, полимиксины и прочие гидрофобные препараты диффундируют сквозь билипидный слой (Kumar, 2005; Alekshun et al., 2007; Delcour, 2009). Было показано, что коровая часть липосахарида (ЛПС), находящегося в верхнем слое ВМ, играет основную роль в барьерной функции ВМ по отношению к гидрофобным антибиотикам, таким как аминогликозиды (гентамицин и канамицин), макролиды (эритромицин), рифамицин, новобиоцин, фузидовая кислота и положительно заряженные пептиды, а также и к тетрациклинам и фторхинолонам, хотя последние два семейства антибиотиков также способны проникать и сквозь порины. Бактериальные штаммы, экспрессирующие длинные олиго- и полисахаридные части ЛПС имеют врожденную устойчивость к этим антибиотикам (Nikaido, 2003; Delcour, 2009). Не менее важным также является устойчивость, контролируемая поринами. Как было указано выше, антибиотики β-лактамного ряда, а также тетрациклины, фторхинолоны и хлорамфеникол проникают во внутреннее пространство клетки через эти мембранные структуры. Поэтому основным механизмом устойчивости к антибиотикам является снижение экспресии генов, кодирующих порины разных типов. Кроме того было показано, что регуляторные изменения экспрессии генов, влекущие за собой и изменение состава поринов, способны привести к колоссальному повышению минимальных ингибирующих концентраций по отношению к таким антибиотикам как цефотаксим и цефокситин. Некоторые виды бактерий, как например Pseudomonasaeruginosa, обладают устойчивостью на уровне проницаемости мембраны в целом благодаря низкому количеству поринов в составе ВМ, что в совокупности с высокоэффективной работой трансмембранных помп, делает этот микроорганизм чрезвычайно устойчивым к широкому спектру антибиотиков (Delcour, 2009). 4.2.4. Другие механизмы устойчивости.К другим механизмам устойчивости можно отнести образование метаболического «шунта». Примером такого механизма может служить устойчивость к ванкомицину у энтерококков. Ванкомицин, в отличие от других антибиотиков, связывается не с ферментом, а с его субстратом. Механизм действия ванкомицина заключается в необратимом связывании с дипептидом D-Ala-D-Ala, входящим в состав UDP-N-ацетилмурамилпентапептида – мономерного предшественника пептидогликана – основного компонента клеточной стенки микроорганизмов. В результате такого связывания нарушается последняя стадия синтеза пептидогликана – включение предшественника в растущую цепь этого полимера и образование поперечных сшивок. Считалось, что из-за такого механизма действия к данному антибиотику не может возникнуть устойчивости. Однако устойчивость к ванкомицину появилась, хотя для этого потребовалось более 30 лет (Nikaido, 2009). У устойчивых штаммов энтерококков обнаруживается модифицированный предшественник, вместо дипептида D-Ala-D-Ala в его состав входит дипептид D-Ala-D-Lac. Аффинность ванкомицина к D-Ala-D-Lac резко снижена. Синтез модифицированного предшественника является результатом активности как минимум 7 генов, объединенных в оперон, входящий в состав транспозона Tn1546. Этот транспозон чаще локализуется на плазмидах, однако встречается и в составе хромосомы. Два из входящих в оперон генов (vanR и vanS) кодируют систему регуляции экспрессии резистентности. Продукт гена vanS является сенсором присутствия ванкомицина в окружающей среде, а продукт гена vanR является регулятором синтеза продуктов генов vanA, vanH и vanX. Экспрессия генов оперона устойчивости индуцируется только после воздействия на клетку ванкомицина (Evers et al., 1996, Courvalin, 2006). Весьма интересный механизм устойчивости к антибиотикам семейства хинолонов за счет имитации молекулы-мишени был обнаружен у Mycobacteriumsmegmatisи Mycobacteriumbovis. У этих двух видов был обнаружен белок семейства «пентапептидных повторов», кодируемый хромосомным геном mfpA (Montero et al., 2001; Hegde et al., 2005). Было показано, что данный белок по окончании фолдинга приобретает структуру, чрезвычайно схожую с двойной спиралью ДНК. Тандемные повторы пяти аминокислот сворачиваются в правозакрученную спираль такой же ширины, что и спираль ДНК. Более того, показано, что суммарный заряд поверхности такой молекулы и спектр поглощения света также практически идентичны ДНК (Khrapunov et al., 2011). Таким образом, MfpA симулирует структуру ДНК и тем самым служит мишенью для фторхинолонов, что защищает клетку от губительного взаимодействия антибиотика с ДНК-гиразным комплексом (Hedge et al., 2005). 4.2.5. Многообразие механизмов устойчивости бактерий к одному антибиотикуНеобходимо особо подчеркнуть, что устойчивость к одному антибиотику может определяться целым рядом различных ферментов и механизмов. Причем довольно часто даже одна клетка обладает различными механизмами устойчивости к одному и тому же антибиотику. Как было показано на примере с устойчивостью к β-лактамам, ряд бактериальных клинических штаммов содержали DD-транспептидазы (пенициллин-связывающие белки) со сниженной, за счёт изменений в их структуре, аффинностью к антибиотику, а также гены, кодирующие белки инактивации этого лекарства, что значительно снижало восприимчивость микроорганизма к антибиотику (Davies, 1994). 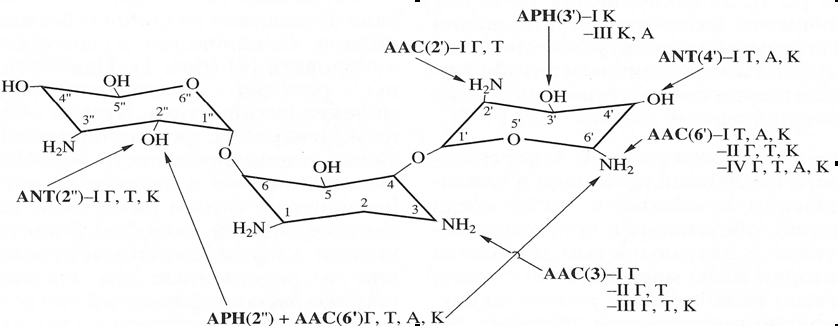 Многообразие способов защиты бактерий от одного и того же антибиотика можно проследить на примере устойчивости к аминогликозидам. Показано, что энзиматическая модификация аминогликозидов является главным способом защиты от этих антибиотиков. При этом способностью модифицировать аминогликозиды обладают ферменты трех классов: 1) N-ацетилтрансферазы (AAC), использующие в качестве донора ацетил-коэнзим А и изменяющие функции аминогруппы; 2) O-нуклеотидилтрансферазы (ANT) и 3) O-фосфотрансферазы (APH), влиящие на функции гидроксильных групп и использующие в качестве донора АТФ (Mingeot-Leclercq,1999). Наиболее распространен механизм ацетилирования с помощью различных ацетилтрансфераз. Ацетилирование может затронуть 1-, 3-, 6’-, и 2’- аминогруппы аминогликозида. Аналогично этому различные гидроксильные группы аминогликозида могут стать мишенью действия О-фосфотрансфераз и О-нуклеотидилтрансфераз. На рис 1. схематически представлены способы энзиматической инактивации канамицина. Видно, что некоторые участки молекулы антибиотика являются мишенью действия нескольких изозимов, обладающих индивидуальной субстратной специфичностью (Mingeot-Leclercq,1999). Таким образом, устойчивость даже к одному антибиотику зависит от действия многих генов и, следовательно, огромное количество генов задействовано в определении устойчивости к антибиотикам, использующимся в различных областях медицины, ветеринарии и сельского хозяйства. Рисунок 1. Действие ферментов, модифицирующих аминогликозиды (на примере канамицина). АРН – фосфотрансфераза; ANT – аденилтрансфераза; ААС –ацетилтрансфераза; APH(2”)+AAC(6’) – бифункциональный фермент; I-IV – изоэнзимы; А-амикацин, К-канамицин, Т-тобрамицин. Составлено на основе Mingeot-Leclercq et al., 1999 (c модификациями). 4.3. Разнообразие генов устойчивости к антибиотикам Общее число генов, кодирующих устойчивость к антибиотикам и описанных к настоящему времени, неизвестно. По-видимому, оно исчисляется несколькими сотнями. Например, одних только генов устойчивости к тетрациклинам было охарактеризовано более 30 (Chopra & Roberts, 2001), а генов, определяющих устойчивость к макролидным антибиотикам (эритромицину, олеандомицину, линкомицину, стрептограмину) – более 40 (Robertset al., 1999). Трудности в определении числа генов устойчивости в первую очередь возникают из-за того, что, практически идентичным генам часто присваивают разные обозначения, в особенности тогда, когда разные группы исследователей независимо выделяют их из разных бактерий. Эта ситуация в последние годы постепенно исправляется благодаря специально проводимым работам по систематизации генов устойчивости различных групп и разработке новой номенклатуры (Robertset al., 1999; Chopra & Roberts, 2001; Shawet al., 1993). В настоящее время в качестве основных методов идентификации генов используют методы ДНК-ДНК гибридизации, ПЦР-амплификации и секвенирования. Разработаны наиболее объективные критерии определения родственных отношений генов и их принадлежности к одному классу (семейству): два или более генов рассматривают в качестве близкородственных, когда их белковые продукты содержат 80% и более идентичных аминокислот и относят к различным классам (семействам), если содержание идентичных аминокислот в белках составляет 79% и менее (Robertset al., 1999; Chopra & Roberts, 2001). Использование этих критериев позволяет идентифицировать как ранее описанные, так и вновь обнаруженные гены клинических и природных штаммов бактерий, а также выявлять случаи горизонтального переноса генов между неродственными бактериями (Werneret al., 2001; Nandiet al., 2004). Следует отметить, что результаты сравнительного анализа структуры большого числа белков, характеризующихся сходным механизмом действия, позволяют сделать вывод об очень древнем происхождении соответствующих генов (Shawet al., 1993). Список цитированной литературы Alekshun M., Levy S., 2007. Molecular Mechanisms of Antibactial Multidrug Resistance. Cell. 128: 1037 – 1050. Biswas S., Raoult D., Rolain J.-M., 2008. A bioinformatic approach to understanding antibiotic resistance in intracellular bacteria through whole genome analysis. Int. J. Antimicrob. Agents 32: 207-220. Chopra I., Roberts M. 2001. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 65: 232-260. Courvalin P. 2006. Vancomycin resistance in gram-positive cocci. Clin Infect Dis. 42 (Suppl 1): S25-34. Davies J. 1994. Inactivation of antibiotics and the dissemination of resistance genes. Science 264: 375-382. Delcour A., 2009. Outer membrane permeability and antibiotic resistance. Biochimica et Biophysica Acta. 1794: 808–816. Evers S., Quintiliani R., Courvalin P. 1996. Genetics of glycopeptide resistance in enterococci. Microb Drug Resist. 2: 219-23.Fernández L., Breidenstein Elena B.M., Hancock Robert E.W. 2011. Creeping baselines and adaptive resistance to antibiotics. Drug Res. Updat. 14: 1 – 21 Hegde S., Vetting M., Roderick S., Mitchenall L., Maxwell A., Takiff H., Blanchard J. 2005. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308: 1480-1483. Hooper D. 2000. Mechanisms of action and resistance of older and newer fluoroquinolones. Clin. Infect. Dis. Suppl 2: S24-28. Khrapunov S., Brenowitz M. 2011. Stability, denaturation and refolding of Mycobacterium tuberculosis MfpA, a DNA mimicking protein that confers antibiotic resistance. Biophys. Chem. 159: 33-40. Kohanski M., Dwyer D., Collins J., 2010. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 8: 423 – 435. Kumar A., Schweizer H., 2005. Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv. Drug Deliv. Rev., 57: 1486 – 1513. Li X.-Z., Nikaido H., 2010. Efflux Mediated Resistance in Bacteria: an Update. Drugs, 69(12): 1555 – 1623. Marquez B., 2005. Bacterial efflux systems and efflux pumps inhibitors. Biochimie 87: 1137 – 1147. Mingeot-Leclercq M.-P., Glupczynski Y., Tulkens P.M. 1999. Aminoglycosides: activity and Resistance. Antimicrob. Agents Chemother. 43: 727-737. Montero C., Mateu G., Rodriguez R., Takiff H. 2001. Intrinsic resistance of Mycobacterium smegmatis to fluoroquinolones may be influenced by new pentapeptide protein MfpA. Antimicrob. Agents Chemother. 45: 3387 – 3392. Nandi S., Maurer J.J., Hofacre C., Summers A.O. 2004. Gram-positive bacteria are a major reservoir of class 1 antibiotic resistance integrons in poultry litter. Proc. Natl. Acad. Sci. USA. 101: 7118-7122. Nikaido H. 2001. Preventing drug access to targets: cell surface permeability barriers and active efflux in bacteria. Seminars in Cell & Developmental Biology 12: 215-223. Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Reviews 67: 593-656. Nikaido H. 2009. Multidrug resistance in bacteria. Annu Rev Biochem. 78: 119-146. Poehlsgaard J., Douthwaite S. 2005. The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 3: 870 – 880. Queenan A., Bush K. 2007. Carbapenemases: the versatile β-lactamases. Clin. Microbiol. Rev. 20: 440-458 Ramirez M., Tolmasky M. 2010. Aminoglycoside modifying enzymes. Drug Res. Updat. 13: 151 – 171. Roberts M., Sutcliffe J., Courvalin P., Jensen L., Rood J., Seppala E. 1999. Nomenclature for macrolide and macrolide-lincosamide-streptogramin B resistance determinants. Antimicrob. Agents Chemother. 43: 2823-2830. Shaw K., Rather P., Hare R., Miller G. 1993. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 57: 138-163. Singh M., Dominy B. 2012. The evolution of cefotaximase activity in the TEM β-lactamase. J. Mol. Biol. 415: 205 – 220. Spratt B. 1994. Resistance to antibiotics mediated by target alterations. Science. 264: 388-393. van Hoek A., Mevius D., Guerra B., Mullany P., Roberts A., Aarts H. 2011. Acquired antibiotic resistance genes: an overview. Front. Microbiol. 2: Аrticle 203. Werner G., Hilderbrandt B., Witte W. 2001. Aminoglycoside-streptothricin resistance gene cluster aadE-sat4-aphA-3 disseminated among multiresistant isolates of Enterococcus faecium. Antimicrob. Agents Chemother. 45: 3267-3269. Wright G., 2005. Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Delivery, 57: 1451 – 1470. Zgurskaya H.I., 2002. Molecular analysis of efflux pump-based antibiotic resistance. Int. J. Med. Microbiol., 292: 95 – 105. |