Реферат Механизмы карбоновых кислот. Отчет по производственной практике Направление подготовки 18. 03. 01 Химическая технология
 Скачать 182.72 Kb. Скачать 182.72 Kb.
|

|
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ Федеральное государственное бюджетное образовательное учреждение высшего образования «Чувашский государственный университет имени И.Н. Ульянова» Химико-фармацевтический факультет Кафедра химической технологии и защиты окружающей среды Отчет по производственной практике Направление подготовки: 18.03.01 – Химическая технология
Чебоксары 2021 Содержание Механизмы реакций замещения карбоновых кислот и их производных с разрывом ацил-кислородной связи 3 Реакционная способность карбоновых кислот и их производных 11 Список источников 15 Механизмы реакций замещения карбоновых кислот и их производных с разрывом ацил-кислородной связиКарбонильная группа карбоновых кислот, как и в случае альдегидов и кетонов, способна взаимодействовать с нуклеофильными реагентами. Однако активность карбоновых кислот в таких реакциях значительно ниже, чем альдегидов и кетонов. Это объясняется по меньшей мере двумя причинами: 1. Гидроксигруппа за счет своего +М-эффекта заметно уменьшает положительный заряд на карбонильном атоме углерода: 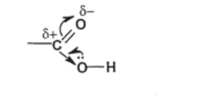  Группа ОН, кроме того, обладает противоположно действующим -I- эффектом, но +М-эффект этой группы намного сильнее, так что в итоге проявляется заметный донорный эффект, понижающий карбонильную активность. 2. Нуклеофилы одновременно являются основаниями; если основность реагента достаточна (а это может быть и основание средней силы), то реагент не атакует карбонильный атом углерода, а просто отрывает протон от группы ОН и образует соль; так, в частности, реагируют аммиак и амины, гидроксил-амин, гидразин, цианид-анион и др.: 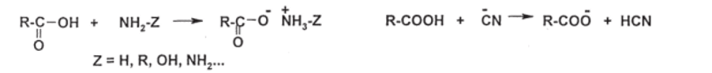 Карбонильная активность в солях карбоновых кислот намного меньше, чем в самих карбоновых кислотах; она почти не проявляется. Причина в том, что анионный фрагмент -О проявляет сильнейший +М-, а также и значительный +I эффекты. Поэтому после образования солей нуклеофильные реагенты в большинстве случаев не реагируют с ними; исключением являются очень сильные нуклеофилы - литий- и магнийорганические соединения и алюмогидрид лития [имеется в виду, конечно, избыток нуклеофильного реагента; его эквивалент, пошедший на образование соли, теряет свои нуклеофильные свойства]. Лишь нуклеофилы, проявляющие слабые основные свойства (например, спирты) не образуют солей; однако они являются также и слабыми нуклеофилами, поэтому без специальной активации они не способны присоединяться по карбонильной группе. Нуклеофильные реакции карбонильной группы карбоновых кислот достаточно эффективно протекают либо при активации реагирующих партнеров (чаще - карбоновой кислоты), либо при высокой температуре, либо с очень сильными нуклеофилами (без активации и в достаточно мягких условиях). После присоединения нуклеофильного реагента следует отщепление гидроксильной группы, и конечным итогом реакции оказывается нуклеофильное замещение группы ОН на фрагмент нуклеофильного реагента по схеме «присоединение - отщепление»: 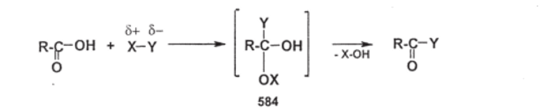 Отщепление происходит потому, что промежуточный продукт присоединения (584) малоустойчив из-за нахождения двух или даже трех электроотрицательных групп у одного атома углерода (три группы в том случае, если группа Y электроотрицательна). Приведенная схема является чисто формальной; конкретные механизмы могут заметно отличаться. Наибольшее число реакций подобного типа приводит к образованию производных карбоновых кислот, сложных эфиров (Y=OR'), амидов (Y= NH2, NHR, NR2), галогенангидридов (Y=Hal), ангидридов (Y=OCOR) и других производных. При взаимодействии с металлорганическими соединениями могут также образовываться кетоны (Y= R1) и продукты их дальнейших превращений. Гидридное восстановление кислот дает первичные спирты. 1. Образование сложных эфиров. Наиболее распространенный вариант образования сложных эфиров из карбоновых кислот - взаимодействие карбоновых кислот со спиртами; в большинстве случаев для этого требуется катализ сильными минеральными кислотами. Реакция называется этерификацией. 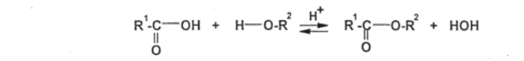 Если в качестве субстрата рассматривать спирт, то она представляет собой ацилирование спирта карбоновой кислотой. Кислотный катализ направлен на активацию либо кислоты (чаще), либо спирта (реже). Известны три механизма взаимодействия карбоновых кислот со спиртами при кислотном катализе. 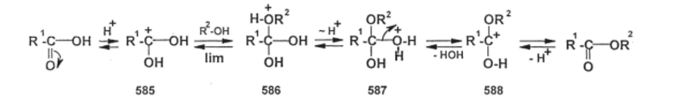 Первый, самый распространенный механизм, можно представить следующим образом: На первой стадии происходит активация карбонильной группы путем ее О-протонирования с образованием интермедиата, который удобно представить резонансной структурой (585); эта стадия является быстрой. Далее следует скоростьопределяющая (лимитирующая) стадия - собственно нуклеофильная атака молекулы спирта на активированную карбонильную группу (как при образовании полуацеталей,). Затем происходит миграция протона в интермедиате (586) от алкокси- к гидроксигруппе, после чего от образовавшегося интермедиата (587) отщепляется вода («хорошая» уходящая группа). Образовавшийся при этом интермедиат (588) - О-протонированный сложный эфир; на заключительной стадии происходит депротонирование и образуется конечный продукт - сложный эфир. Все стадии данного превращения обратимы (реакция микроскопически обратима); поэтому в присутствии водных растворов кислот сложные эфиры гидролизуются с образованием исходных кислот и спиртов по тому же механизму, проходящему в обратном направлении (кислотный гидролиз сложных эфиров). Обратимость реакции этерификации по данному механизму выражена весьма заметно, поэтому для того, чтобы сдвинуть равновесие в сторону образования сложного эфира, необходимо удалять образующуюся воду. Лимитирующая стадия данного механизма бимолекулярна, т.е. и сама реакция является бимолекулярной. Второй механизм встречается значительно реже - в случае пространственно затрудненных кислот, таких, как о-дизамещенные ароматические карбоновые кислоты. Реакция по бимолекулярному механизму в этом случае затруднена, т.к. ключевой интермедиат (586) пространственно напряжен (тетракоординированный атом углерода). В этом случае реакцию проводят в сильнокислой среде: например, растворяют карбоновую кислоту в конц. H2SO4 и осторожное выливают раствор в спирт; механизм при этом следующий: 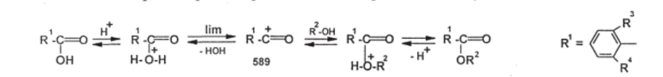 После О-протонироваиия гидроксигруппы на скоростьопределяющей стадии образуется ключевой интермедиат - ацил-катион (ацилий-катион) (589); в этом интермедиате атом углерода двухкоординирован, поэтому даже самый разветвленный радикал R1 не создает пространственного напряжения. Далее (после выливания в спирт) следует нуклеофильная атака молекулы спирта, и после депротонирования образуется конечный продукт. Лимитирующая стадия мономолекулярна, этот механизм обозначают как Аас1. Реакция и здесь микроскопически обратима; подобным образом проводят кислотный гидролиз пространственно затрудненных эфиров. Третий механизм имеет место только в тех случаях, когда спирт R2OH может образовать достаточно устойчивый карбокатион (третичные спирты, спирты с бензильными или аллильными радикалами). В этом случае кислотный катализ направлен не на карбоновую кислоту, а на спирт, из которого генерируется карбокатион (590):  Данный механизм - кислотно-катализируемое SNJ- замещение группы ОН в спиртах: роль нуклеофила выполняет карбоновая кислота. Реакцию обозначают как Aal1 (А1 - алкил); действительно, в двух предыдущих механизмах разрывается связь О-ацил, а в данном - связь О-алкил. В механизмах Аас2 и Аас1 нуклеофилом является спирт, электрофилом - карбоновая кислота; в механизме Aal1 ситуация противоположная. Иногда вместо минеральных кислот в качестве катализатора этерификации используют кислоту Льюиса - эфират трифторида бора. Некоторые, относительно сильные кислоты (муравьиная, щавелевая) могут этерифицироваться и в отсутствие катализатора. Для получения сложных эфиров может использоваться взаимодействие солей карбоновых кислот с галогенпроизводными, тозилатами и другие соединениями с хорошими нуклеофильными группами: 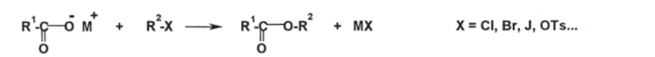 Это - обычные реакции нуклеофильного замещения. Образование амидов. Как уже упоминалось, карбоновые кислоты при взаимодействии с аммиаком образуют соли. При нагревании этих солей до достаточно высокой температуры (выше 150 °С) происходит их дегидратация с образованием амидов:  Вначале аммонийная соль диссоциирует на исходные карбоновую кислоту и аммиак, а затем аммиак нуклеофильно присоединяется по карбонильной группе кислоты: 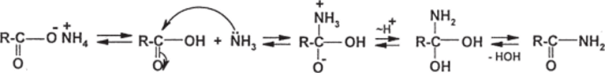 Присоединение становится возможным благодаря жестким условиям; к тому же аммиак заметно более сильный нуклеофил, чем спирты. Образование галогенангидридов (ацилгалогенидов). Для замещения карбоксильной гидроксигруппы на галоген обычно используют уже знакомые реагенты - галогениды фосфора или тионилхлорид: 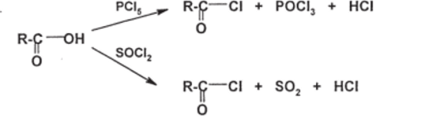 Замещению предшествует активация гидроксигруппы: 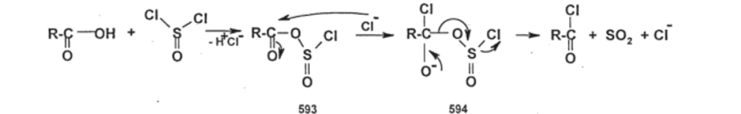 Галогенид-ион атакует «активированный субстрат» (593); этому способствует увеличение частичного положительного заряда на атоме углерода за счет акцепторного влияния фрагмента SOC1; в образовавшемся интермедиате (594) далее происходит «внутреннее нуклеофильное замещение»; этому благоприятствует «хорошая» уходящая группа OSOC1. Среди галогенангидридов наиболее широко используются хлорангидриды, как наиболее доступные. Иногда используют бромангидриды; для их получения применяют РВr3 или SOBr2. Производные карбоновых кислот, как и сами кислоты, способны вступать в реакции нуклеофильного замещения у sp2-гибридизованного атома углерода с образованием других функциональных производных. Тетраэдрический механизм нуклеофильного замещения. Сначала нуклеофил присоединяется к атому углерода группы С=О с образованием нестабильного промежуточного аниона (интермедиата). Механизм реакции называют тетраэдрическим, так как атом углерода при этом переходит из sp2- в sр3-гибридное состояние и принимает тетраэдрическую конфигурацию. 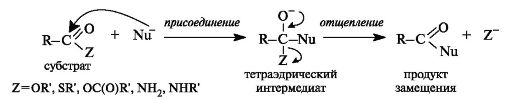 На второй стадии от интермедиата отщепляется частица Z и атом углерода вновь становится sp2-гибридизованным. Таким образом, эта реакция замещения включает стадии присоединения и отщепления. По такому механизму реакция протекает при наличии достаточно сильного нуклеофила и хорошей уходящей группы Z, например, в случае щелочного гидролиза сложных эфиров и других функциональных производных карбоновых кислот. Легкость нуклеофильной атаки зависит от величины частичного положительного заряда δ+ на атоме углерода карбонильной группы. В функциональных производных карбоновых кислот он увеличивается с ростом -I-эффекта заместителя Z и уменьшается с увеличением его M-эффекта. В результате этих эффектов величина заряда и, следовательно, способность подвергаться нуклеофильной атаке в рассматриваемых соединениях уменьшаются в приведенной ниже последовательности. К этому же выводу приводит и анализ стабильности уходящих групп Z-.
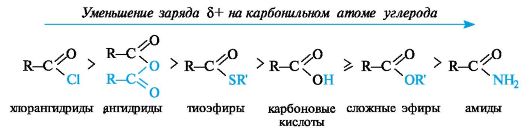 Производные карбоновых кислот по сравнению с альдегидами и кетонами труднее подвергаются нуклеофильной атаке, так как электрофильность карбонильного атома углерода обычно снижается за счет +M-эффекта заместителя Z. По этой причине в нуклеофильных реакциях функциональных производных карбоновых кислот часто оказывается необходимым кислотный катализ путем протонирования атома кислорода карбонильной группы. В результате взаимодействия карбоновых кислот и их функциональных производных со спиртами или аминами в молекулы этих соединений вводится ацильный остаток. По отношению к таким реакциям используют общее название - реакции ацилирования. С этой позиции реакцию этерификации можно рассматривать как ацилирование молекулы спирта. Реакционная способность карбоновых кислот и их производныхХимические свойства карбоновых кислот почти полностью определяются карбоксильной группой -СО-ОН - функциональной группой, в которой карбонильная и гидроксильная группы непосредственно связаны и поэтому заметно влияют друг на друга. Вследствие этого карбоновые кислоты, сохраняя определенное сходство с карбонильными соединениями и со спиртами, приобретают весьма заметное своеобразие. Это своеобразие касается, прежде всего, двух аспектов: А. По сравнению со спиртами и фенолами карбоновые кислоты проявляют гораздо более сильные кислотные свойства. Б. По сравнению с альдегидами и кетонами карбоновые кислоты труднее присоединяют нуклеофильные реагенты; в большинстве случаев после присоединения происходит отщепление гидроксильной группы. Карбоксильная группа, подобно некоторым ранее рассмотренным группам, активирует связанное с ней а-положение. Химические свойства карбоновых кислот можно разделить на 4 группы: I. Реакции с разрывом связи О-Н (кислотные свойства); II. Реакции по карбонильной группе (взаимодействие с нуклеофилами); III. Реакции, сопровождающиеся потерей карбоксильной группы (декарбоксилирование и декарбонилирование); IV. Реакции a-положения к карбоксильной группе. 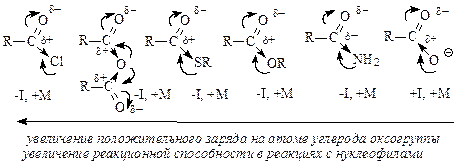 Величина положительного заряда, возникающего на атоме углерода оксогруппы, увеличивается с усилением электроноакцепторного характера заместителя (d+ возрастает с усилением отрицательного индуктивного эффекта по сравнению с положительным мезомерным эффектом). Отличием приведенных выше соединений от альдегидов и кетонов является то, что 2 электрона p-связи углерод-кислород участвуют в сопряжении с неподеленной парой р-электронов гетероатома (атомов галогена, серы, кислорода или азота), что приводит к повышению стабильности соединений. Нуклеофильное присоединение к карбонильной группе этих соединений приводило бы к изменению гибридизации атомных орбиталей углерода с sp2 на sр3 и разрушению системы сопряжения, уменьшению стабильности молекулы. Для сохранения сопряженной системы в реакциях с нуклеофилами после присоединения нуклеофильной частицы происходит отщепление уходящей группы и регенерация сопряжения. В итоге нуклеофильная частица оказывается связанной с ацильной группой – R-C=O, поэтому реакции производных карбоновых кислот называют реакциями ацилирования. Общая схема нуклеофильного замещения имеет вид: 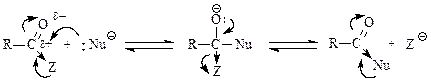 Реакционная способность соединений зависит от величины эффективного положительного заряда на атоме углерода карбонильной группы и от способности к отщеплению уходящей группы Z. В приведенном выше ряду функциональных производных карбоновых кислот справа налево (от солей к галогенангидридам) увеличивается положительный заряд на атоме углерода и увеличивается стабильность уходящей группы, т.е. способность к отщеплению. Ангидриды карбоновых кислот несколько менее реакционно способны в реакциях ацилирования вследствие меньшего положительного заряда на атомах углерода карбонильных групп. Меньший положительный заряд в сравнении с галогенангидридами возникает из-за того, что атом кислорода проявляет отрицательный индуктивный эффект по отношению к обеим карбонильным группам. При ацилировании спиртов ангидридами кислот, как и при ацилировании с помощью галогенангидридов, выделяющуюся кислоту связывают добавлением органических оснований. Карбоновые кислоты и сложные эфиры карбоновых кислот являются существенно менее реакционноспособными ацилирующими агентами. Снижение реакционной способности обусловлено снижением эффективного положительного заряда на атоме углерода карбонильной группы из-за усиления электронодонорных свойств гидроксильной группы ОН у кислот или алкоксигруппы ОR у сложных эфиров в сравнении с ацильной группой или атомом галогена. Карбоновые кислоты, кроме электрофильного центра, имеют и другие реакционные центры: 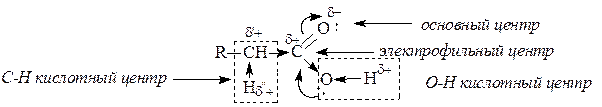 В отличие от альдегидов и кетонов, карбоновые кислоты имеют О-Н кислотный центр, за счет которого проявляют сравнительно сильные кислотные свойства. Сила карбоновых кислот зависит от величины эффективного положительного заряда на атоме углерода карбоксильной группы. Чем больше величина этого заряда, тем сильнее пара электронов атома кислорода гидроксильной группы вовлечена в р,p-сопряжение, тем сильнее поляризована связь и ослаблена связь О-Н, тем легче идет ее разрыв с образованием мезомерно стабилизированного карбоксилат-аниона:  Именно из-за стабильности карбоксилат-анионов и легкости их образования карбоновые кислоты проявляют более сильные кислотные свойства, чем фенолы, тиолы и спирты. Величина эффективного заряда на атоме углерода карбоксильной группы и кислотные свойства зависят от характера радикала, связанного с карбоксильной группой: электроноакцепторные заместители усиливают кислотные свойства соединений, электронодонорные – ослабляют.  Реакционная способность карбоновых кислот в реакциях нуклеофильного замещения в сравнении с галогенангидридами, ангидридами, тиоэфирами понижена вследствие электронодонорных свойств ОН-группы, уменьшающей электрофильность атома углерода. Для увеличения электрофильности (эффективного положительного заряда) необходимо использовать кислотный катализ. Список источниковБелобородов В.Л., Зурабян С.Э., Лузин А.П., Тюкавкина Н.А. Органическая химия. Основной курс. – М.: Дрофа, 2003. Каминский, В. А. Органическая химия в 2 ч. Часть 2: учебник для среднего профессионального образования / В. А. Каминский. — 2-е изд., испр. и доп. - Москва: Издательство Юрайт, 2020. Неницеску К.Д. Органическая химия. В 2 т. – М.: Изд-во иностранной литературы, 1963. Несмеянов А.Н., Несмеянов Н.А. Начала органической химии. В 2 т. – М.: Химия, 1969. Травень В.Ф. Органическая химия. Учебник для вузов. – М.: Академкнига, 2005. |