Программы биоинформатики. ClustalW и другие программы множест№выр. Отображает шаги, которые алгоритм программного обеспечения ClustalW использует для глобального выравнивания
 Скачать 88.74 Kb. Скачать 88.74 Kb.
|

|
ClustalW 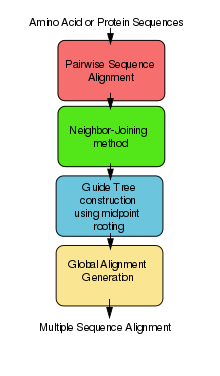 Отображает шаги, которые алгоритм программного обеспечения ClustalW использует для глобального выравнивания ClustalW, как и другие инструменты Clustal, используется для эффективного выравнивания множественных нуклеотидных или белковых последовательностей. Он использует методы прогрессивного выравнивания, которые сначала выравнивают наиболее похожие последовательности и постепенно переходят к наименее похожим последовательностям, пока не будет создано глобальное выравнивание. ClustalW - это матричный алгоритм, тогда как такие инструменты, как T-Coffee и Dialign, основаны на согласованности. ClustalW имеет довольно эффективный алгоритм, который хорошо конкурирует с другим программным обеспечением. Этой программе требуются три или более последовательностей для вычисления глобального выравнивания, для попарного выравнивания последовательностей (2 последовательности) используйте инструменты, подобные EMBOSS, LALIGN.  Диаграмма, показывающая метод соединения соседей в последовательности выравнивание для биоинформатики Диаграмма, показывающая метод соединения соседей в последовательности выравнивание для биоинформатикиАлгоритм ClustalW использует методы прогрессивного выравнивания, как указано выше. В них сначала выравниваются последовательности с наилучшей оценкой выравнивания, затем выравниваются постепенно более отдаленные группы последовательностей. Этот эвристический подход необходим из-за того, что для поиска глобального оптимального решения требуется время и память. Первым шагом алгоритма является вычисление приблизительной матрицы расстояний между каждой парой последовательностей, также известной как попарное выравнивание последовательностей. Следующим шагом является метод соединения соседей, который использует укоренение средней точки для создания общего направляющего дерева. Процесс, который он использует для этого, показан на подробной диаграмме метода справа. Затем дерево направляющих используется в качестве приблизительного шаблона для генерации глобального выравнивания. Временная сложность ClustalW имеет временную сложность O (N 2) {\ displaystyle O (N ^ {2})}из-за его использования метода объединения соседей. В обновленной версии (ClustalW2) в программное обеспечение встроена опция для использования UPGMA, что быстрее при больших размерах ввода. Флаг командной строки, позволяющий использовать его вместо объединения соседей: -clustering = UPGMA Например, на стандартном рабочем столе выполнение UPGMA для 10 000 последовательностей даст результаты менее чем за минуту, в то время как сосед- присоединение заняло бы больше часа. Запуск алгоритма ClustalW с этой настройкой позволяет значительно сэкономить время. ClustalW2 также имеет возможность использовать итеративное выравнивание для повышения точности выравнивания. Хотя это не обязательно быстрее или эффективнее с точки зрения сложности, повышение точности ценно и может быть полезно для данных меньшего размера. Для этого используются различные флаги командной строки: -Iteration = Alignment -Iteration = Tree -numiters Первая опция командной строки уточняет окончательное выравнивание. Второй вариант включает схему в шаг последовательного выравнивания алгоритма. Третий определяет количество циклов итераций, где значение по умолчанию установлено на 3. По сути, Clustal создает несколько выравниваний последовательностей с помощью трех основных шагов: Выполните попарное выравнивание, используя метод прогрессивного выравнивания Создайте направляющее дерево (или используйте определяемое пользователем дерево) Используйте направляющее дерево для выполнения множественного выравнивания Эти шаги выполняются автоматически, когда вы выбираете «Выполнить полное выравнивание». Другие варианты: «Выполнить выравнивание по дереву направляющих и филогении» и «Создать только дерево направляющих». MUSCLE Сравнение нескольких последовательностей по протоколу -Expectation () - компьютерное программное обеспечение для множественного выравнивания последовательностей последовательностей белков и нуклеотидов.. Он под лицензией как общественное достояние. Этот метод был опубликован Робертом С. Эдгаром в двух статьях в 2004 году. В первой статье, опубликованной в Nucleic Acids Research, был представлен алгоритм выравнивания последовательностей. Во второй статье, опубликованной в BMC Bioinformatics, представлены более технические детали. Алгоритм Алгоритм MUSCLE состоит из трех этапов: предварительный прогрессивный, улучшенный прогрессивный и этап уточнения. На этапе прогрессивного наброска алгоритм производит предварительное множественное выравнивание, делая упор на скорость важнее точности. На улучшенной прогрессивной стадии расстояние Кимуры используется для переоценки бинарного дерева для создания чернового совмещения, что, в свою очередь, обеспечивает более точное множественное совмещение. На заключительном этапе уточнения уточняется улучшенное выравнивание, выполненное на втором этапе. В конце каждого этапа доступно несколько выравниваний. На первых двух этапах алгоритма временная сложность равна O (NL + NL), пространственная сложность равна O (N + NL + L). Этап уточнения добавляет к временной сложности еще один член, O (NL). MUSCLE часто используется вместо Clustal, поскольку обычно (но не всегда) дает лучшее выравнивание последовательностей в зависимости от выбранных параметров. Кроме того, MUSCLE значительно быстрее, чем Clustal, в большей степени для больших выравниваний. Интеграция MUSCLE интегрирована в программное обеспечение Lasergene от DNASTAR, Geneious и MacVector и доступен в Sequencher, MEGA и UGENE в виде плагина. MUSCLE также доступен в виде веб-службы через Европейскую лабораторию молекулярной биологии (EMBL) - Европейский институт биоинформатики (EBI). По состоянию на сентябрь 2016 года две статьи, описывающие MUSCLE, были процитированы более 19000 раз. MAFFT В биоинформатике, MAFFT (для m ultiple a lignment using f ast F ourier t ransform) - программа, используемая для создания множественных выравниваний последовательностей из аминокислоты или нуклеотидные последовательности. Опубликованная в 2002 году первая версия MAFFT использовала алгоритм, основанный на прогрессивном выравнивании, в котором последовательности были кластеризованы с помощью быстрого преобразования Фурье. В последующих версиях MAFFT были добавлены другие алгоритмы и режимы работы, включая опции для более быстрого выравнивания большого количества последовательностей, выравнивания с более высокой точностью, выравнивания некодирующих последовательностей РНК и добавления новых последовательностей к существующим. Репозиторий; mafft.cbrc.jp / alignment / software / source.html 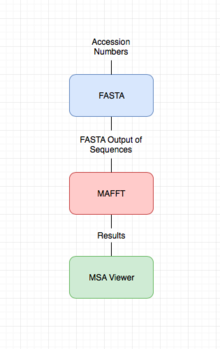 Шаги по использованию MAFFT с другими программами для просмотра MSA T-Coffee Другой распространенный метод прогрессивного выравнивания называется T-Coffee [16] . , работает медленнее, чем Clustal и его производные, но обычно обеспечивает более точное выравнивание для отдаленно связанных наборов последовательностей. T-Coffee вычисляет попарное выравнивание, комбинируя прямое выравнивание пары с непрямым выравниванием, которое выравнивает каждую последовательность пары с третьей последовательностью. Он использует выходные данные Clustal, а также другой программы локального выравнивания LALIGN, которая находит несколько областей локального выравнивания между двумя последовательностями. Полученное выравнивание и филогенетическое дерево используются в качестве ориентира для получения новых и более точных весовых коэффициентов. Поскольку прогрессивные методы представляют собой эвристические методы, сходимость которых к глобальному оптимуму не гарантируется, качество выравнивания может быть трудно оценить, а их истинное биологическое значение может быть неясным. В программе PSAlign реализован полупрогрессивный метод, который улучшает качество выравнивания и не использует эвристику с потерями, но при этом работает за полиномиальное время |