Патологическая анатомия
 Скачать 2.84 Mb. Скачать 2.84 Mb.
|

ЭтиологияНа данный момент единой теории возникновения данного заболевания нет. Выдвигаются следующие варианты, а также их сочетания: теория липопротеидной инфильтрации — первично накопление липопротеидов в сосудистой стенке, теория дисфункции эндотелия — первично нарушение защитных свойств эндотелия и его медиаторов, аутоиммунная — первично нарушение функции макрофагов и лейкоцитов, инфильтрация ими сосудистой стенки, моноклональная — первично возникновение патологического клона гладкомышечных клеток, вирусная — первично вирусное повреждение эндотелия (герпес, цитомегаловирус и др.), перекисная — первично нарушение антиоксидантной системы, генетическая — первичен наследственный дефект сосудистой стенки, хламидиозная — первичное поражение сосудистой стенки хламидиями, в основном, Chlamydia pneumoniae. гормональная — возрастное повышение уровня гонадотропных и адренокортикотропных гормонов приводит к повышеному синтезу строительного материала для гормонов-холестерина. Факторы рискакурение (наиболее опасный фактор) гиперлипопротеинемия (общий холестерин > 5 ммоль/л, ЛПНП > 3 ммоль/л, ЛП(a) > 50 мг/дл) артериальная гипертензия (систолическое АД > 140 мм рт. ст. диастолическое АД > 90 мм рт. ст.) сахарный диабет ожирение малоподвижный образ жизни (гиподинамия) эмоциональное перенапряжение неправильное питание наследственная предрасположенность постменопауза гиперфибриногенемия гомоцистеинурия гипотиреоз[2] Согласно Европейскому руководству по предотвращению сердечно-сосудистых заболеваний (European Guidelines on Cardiovascular Disease Prevention) оценка ведущих факторов риска проводится на основании шкалы SCORE (Systemic COronary Risk Evaluation). Сейчас на сайте Европейского кардиологического общества (ESC) доступна для использования русифицированная программа расчёта риска развития сердечно-сосудистых заболеваний. Более общая оценка может быть проведена по приведённой таблице. Патогенез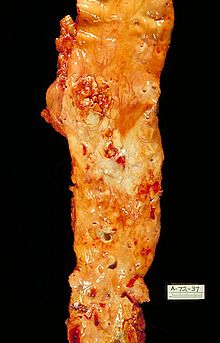 Атеросклеротическое поражение аорты. Патогенез атеросклероза называют атерогенезом. Он происходит в несколько этапов. Развитие атеросклеротического поражения — это совокупность процессов поступления в интиму и выхода из неё липопротеидов и лейкоцитов, пролиферации и гибели клеток, образования и перестройки межклеточного вещества, а также разрастания сосудов и обызвествления. Эти процессы управляются множеством сигналов, часто разнонаправленных. Накапливается всё больше данных о сложной патогенетической связи между изменением функции клеток сосудистой стенки и мигрировавших в неё лейкоцитов и факторами риска атеросклероза. Накопление и модификация липопротеидовВ норме интима артерий образована одноклеточным эндотелиальным слоем, под которым находятся гладкомышечные клетки, погруженные в межклеточное вещество. Первые проявления болезни — так называемые липидные пятна. Их появление связано с местным отложением липопротеидов в интиме. Атерогенными свойствами обладают не все липопротеиды, а только низкой (ЛПНП) и очень низкой плотности (ЛПОНП). Изначально они накапливаются в интиме преимущественно за счёт связывания с компонентами межклеточного вещества — протеогликанами. В местах образования липидных пятен большую роль играет преобладание гепарансульфатов над двумя другими гликозаминогликанами — кератансульфатами и хондроитинсульфатами. В интиме липопротеиды, особенно связанные с протеогликанами, могут вступать в химические реакции. Основную роль играют две: окисление и неферментативное гликозилирование. В интиме в отличие от плазмы содержится мало антиоксидантов. Образуется смесь окисленных ЛПНП, причём окисляются как липиды, так и белковый компонент. При окислении липидов образуются гидроперекиси, лизофосфолипиды, оксистерины и альдегиды (при перекисном окислении жирных кислот). Окисление апопротеинов ведёт к разрыву пептидных связей и соединению боковых цепей аминокислот (обычно β-аминогруппы лизина) с продуктами расщепления жирных кислот (4-гидроксиноненалем и малоновым диальдегидом). Стойкая гипергликемия при сахарном диабете способствует неферментативному гликозилированию апопротеинов и собственных белков интимы, что тоже нарушает их функции и ускоряет атерогенез. Миграция лейкоцитов и образование ксантомных (пенистых) клеток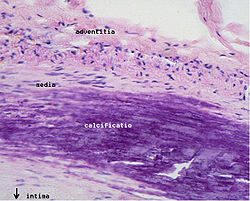 Кальцификация стенки сосуда Миграция лейкоцитов, в основном моноцитов и лимфоцитов, — вторая стадия развития липидного пятна. Их миграцию в интиму обеспечивают расположенные на эндотелии рецепторы — молекулы адгезии. Особого внимания заслуживают молекулы VCAM-1 и ICAM-1 (из суперсемейства иммуноглобулинов) и Р-селектины. Синтез молекул адгезии могут увеличивать цитокины. Так, интерлейкин-1 (ИЛ-1) и фактор некроза опухолей (ФНОα) вызывают или усиливают синтез эндотелиальными клетками VCAM-1 и ICAM-1. В свою очередь, выброс цитокинов клетками сосудистой стенки стимулируется модифицированными липопротеидами. Образуется порочный круг. Играет роль и характер тока крови. В большинстве участков неизмененной артерии кровь течет ламинарно, и возникающие при этом силы снижают экспрессию (проявление) на поверхности эндотелиальных клеток молекул адгезии. Также ламинарный кровоток способствует образованию в эндотелии окиси азота NO. Кроме сосудорасширяющего действия, в низкой концентрации, поддерживаемой эндотелием, NO обладает противовоспалительной активностью, снижая, например, синтез VCAM-1. Но в местах ветвления ламинарный ток часто нарушен, именно там обычно возникают атеросклеротические бляшки. После адгезии лейкоциты проходят через эндотелий и попадают в интиму. Липопротеиды могут непосредственно усиливать миграцию: окисленные ЛПНП способствуют хемотаксису лейкоцитов. К дальнейшему образованию липидного пятна причастны моноциты. В интиме моноциты становятся макрофагами, из которых за счёт опосредованного рецепторами эндоцитоза липопротеидов возникают заполненные липидами ксантомные (пенистые) клетки. Раньше предполагали, что в эндоцитозе участвуют хорошо известные рецепторы ЛПНП, но при дефекте этих рецепторов как у экспериментальных животных, так и у больных (например, при семейной гиперхолестеринемии) всё равно имеются многочисленные ксантомы и атеросклеротические бляшки, заполненные ксантомными клетками. Кроме того, экзогенный холестерин тормозит синтез этих рецепторов, и при гиперхолестеринемии их мало. Теперь предполагается роль скэвенджер-рецепторов макрофагов (связывающих в основном модифицированные липопротеиды) и других рецепторов для окисленных ЛПНП и мелких атерогенных ЛПОНП. Некоторые ксантомные клетки, поглотившие липопротеиды из межклеточного вещества, покидают стенку артерии, препятствуя тем самым накоплению в ней липидов. Если же поступление липопротеидов в интиму преобладает над их выведением с макрофагами (или другими путями), липиды накапливаются и в итоге образуется атеросклеротическая бляшка. В растущей бляшке некоторые ксантомные клетки подвергаются апоптозу или некрозу. В результате в центре бляшки образуется полость, заполненная богатыми липидными массами, что характерно для поздних стадий атерогенеза. Про- и антиатерогенные факторыПри поглощении модифицированных липопротеидов макрофаги выделяют цитокины и факторы роста, способствующие развитию бляшки. Одни цитокины и факторы роста стимулируют деление гладкомышечных клеток и синтез межклеточного вещества, которое накапливается в бляшке. Другие цитокины, особенно интерферон-γ из активированных Т-лимфоцитов, тормозят деление гладкомышечных клеток и синтез коллагена. Такие факторы, как ИЛ-1 и ФНОα, вызывают выработку в интиме тромбоцитарного фактора роста и фактора роста фибробластов, которые играют роль в дальнейшей судьбе бляшки. Таким образом, происходит сложное взаимодействие факторов, как ускоряющих, так и тормозящих атерогенез. Велика роль и небелковых медиаторов. Активированные макрофаги и клетки сосудистой стенки (эндотелиальные и гладкомышечные) вырабатывают свободные радикалы кислорода, которые стимулируют пролиферацию гладкомышечных клеток, усиливают синтез цитокинов, а также связывают NO. С другой стороны, активированные макрофаги способны к синтезу индуцируемой NO-синтазы. Этот высокоактивный фермент вырабатывает NO в высоких, потенциально токсичных концентрациях — в отличие от небольшой концентрации NO, создаваемой конститутивной формой фермента — эндотелиальной NO-синтазой. Помимо макрофагов, в удалении холестерина из поражённой интимы участвуют липопротеиды высокой плотности (ЛПВП), обеспечивающие так называемый обратный транспорт холестерина. Доказана чёткая обратная зависимость между концентрацией холестерина ЛПВП и риском ИБС. У женщин детородного возраста концентрация холестерина ЛПВП выше, чем у сверстников-мужчин, и во многом благодаря этому женщины реже страдают атеросклерозом. В эксперименте показано, что ЛПВП способны удалять холестерин из ксантомных клеток. Участие гладкомышечных клетокАтеросклеротическая бляшка развивается из липидного пятна, но не все пятна становятся бляшками. Если для липидных пятен характерно накопление ксантомных клеток, то для бляшек — фиброз. Межклеточное вещество в бляшке синтезируют в основном гладкомышечные клетки, миграция и пролиферация которых — вероятно, критический момент в образовании фиброзной бляшки на месте скопления ксантомных клеток. Миграцию в липидное пятно гладкомышечных клеток, их пролиферацию и синтез межклеточного вещества вызывают цитокины и факторы роста, выделяемые под влиянием модифицированных липопротеидов и других веществ макрофагами и клетками сосудистой стенки. Так, тромбоцитарный фактор роста, выделяемый активированными эндотелиальными клетками, стимулирует миграцию гладкомышечных клеток из медии в интиму. Образуемые локально факторы роста вызывают деление как собственных гладкомышечных клеток интимы, так и клеток, пришедших из медии. Один из мощных стимуляторов синтеза этими клетками коллагена — трансформирующий фактор роста р. Кроме паракринной (факторы поступают от соседних клеток) происходит и аутокринная (фактор вырабатывается самой клеткой) регуляция гладкомышечных клеток. В результате происходящих с ними изменений ускоряется переход липидного пятна в атеросклеротическую бляшку, содержащую много гладкомышечных клеток и межклеточного вещества. Как и макрофаги, эти клетки могут вступать в апоптоз: его вызывают цитокины, способствующие развитию атеросклероза. Развитие осложнённой бляшкиКроме обычных факторов риска и описанных выше цитокинов на поздних стадиях развития атеросклероза важная роль принадлежит изменениям в свертывающей системе крови. Для появления липидных пятен не требуется повреждения или слущивания эндотелия. Но в дальнейшем в нём могут возникать микроскопические разрывы. На обнаженной базальной мембране происходит адгезия тромбоцитов, и в этих местах образуются мелкие тромбоцитарные тромбы. Активированные тромбоциты выделяют ряд веществ, ускоряющих фиброз. Кроме тромбоцитарного фактора роста и трансформирующего фактора роста р на гладкомышечные клетки действуют низкомолекулярные медиаторы, например серотонин. Обычно эти тромбы растворяются, не вызывая никаких симптомов, и целость эндотелия восстанавливается. По мере развития бляшки в неё начинают обильно врастать vasa vasorum (сосуды сосудов). Новые сосуды влияют на судьбу бляшки несколькими путями. Они создают обширную поверхность для миграции лейкоцитов как внутрь бляшки, так и из неё. Кроме того, новые сосуды — источник кровоизлияния в бляшку: как и при диабетической ретинопатии, они ломкие и склонны к разрыву. Возникающее кровоизлияние ведет к тромбозу, появляется тромбин. Он не только участвует в гемостазе, но и влияет на клетки интимы: стимулирует деление гладкомышечных клеток и выработку ими цитокинов, а также вызывает синтез эндотелием факторов роста. В результате кровоизлияний бляшки часто содержат фибрин и гемосидерин. Атеросклеротические бляшки часто обызвествляются. В бляшках содержатся кальцийсвязывающие белки остеокальцин и остеопонтин и некоторые другие белки, характерные для костной ткани (в частности, белки — регуляторы морфогенеза кости). Гангрена правой нижней конечности вследствие тромбоза бедренной артерии на фоне атеросклероза |