Пептид если от 10 до 40 аминокислот полипептид
 Скачать 7.45 Mb. Скачать 7.45 Mb.
|

47 вопросОбмен фенилаланина и тирозина в тканях. Использование тирозина для синтеза катехоламинов, тироксина, меланина. Распад тирозина до фумарата и ацетоацетата. Наследственные нарушения обмена фенилаланина и тирозина. Фениаланин-незаменимая аминокислота,т.к. в клетках животных не синтезируется её бензольное кольцо. Основное кол-во фенилаланина расходуется по 2-м путям: включается в белки и превращается в тирозин. Превращение фенилаланина в тирозин прежде всего необходимо для удаления избытка фенилаланина,т.к. высокие концентрации его токсичны для клеток. Образование тирозина не имеет большого значения, т.к. недостатка этой аминокислоты в клетках практически не бывает.Основной путь метаболизма фенилаланина начинается с его гидроксилирования в результате чего образуется тирозин. Эта реакция катализируется специфической монооксигеназой-фенилаланингидроксилазой,коферментом которой служит тетрагидробиоптерин Н4БП. Реакция необратима.Н4БП окисляется в Н2БП.  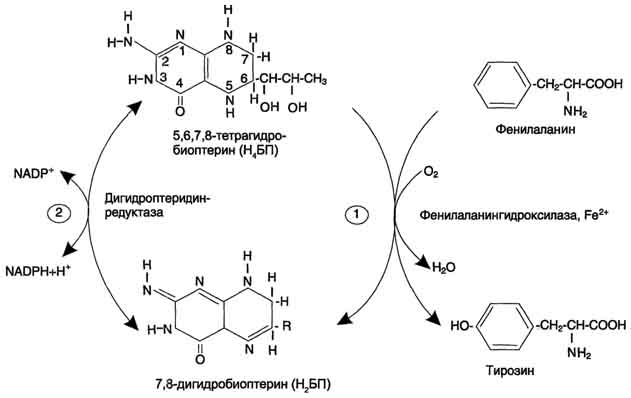 Тирозин-условно заменимая аминокислота, поскольку образуется из фенилаланина. Тирозин в разных тканях выступает предшественником катехоламины,тироксин,меланины и катаболизируется до СО2 и Н2О. В печени происходит катаболизм тирозина до конечных продуктов. Обмен фенилаланина и тирозина связан со значительным кол-ом реакций гидроксилирования,которые катализируют оксигеназы. Трансаминирование тирозина с а-кетоглуторатом катализирует тирозинаминотрасфера. Реакцию окисления в гомог.кислоту катализирует фермент п-гидроксифенилпируватдиоксигеназа. Расщепление аром.кольца катализируется диоксигеназой гомогентизиновой кислоты. Его гидролиз под действием фумарилацетоацетатгидролазы. фумарат может окисляться до СО2 и Н2О,далее глюконеогенез. Ацетоацетат – кетоновое тело, до конечных продуктов в выделением тепла. 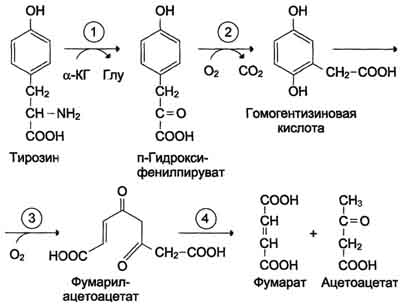 При образовании катехоламинов,которое происходит в нервной ткани и надпочечниках, и меланина в меланоцитах промежуточным продуктом служит диоксифенилаланин(ДОФА) Однако гидроксилирование тирозина в клетках различных типов катализируется различными ферментами: тирозиназа,тирозингидроксилаза. В пигментных клетках тирозин выступает предшественником меланинов. Гормоны тироксин и трийодтиронин прдеставялют собой остатки тирозина, кот попадают в кл щитовидки через БМ. 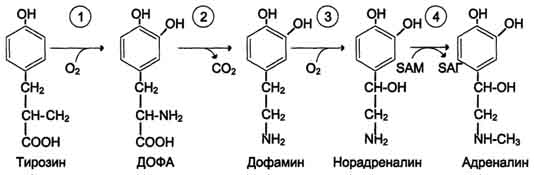 Заболевание-фенилкетонурия. В печени здоровых людей небольшая часть фенилаланина превращается в фениллактат и фенилацетилглутамин. Этот путь катаболизма фенилаланина становится главным при нарушении основного пути-превращения в тирозин,катализируемого фенил-аланингидроксилазой.Повышается содержание фенилпирувата, фенилацетата, фениллактата, фенилацетилглутамина. Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия(ФКУ).выделяют Классическая(наследственное, мутации в гене фенилаланингидроксилазы) и вариантная(коферментзависимая, мутации в геназ, котрол-х метаболизм Н4БП) ФКУ. Наиболее тяжёлые проявления ФКУ-нарушение умственного и физического развития,судорожный синдром,нарушение пигментации. Тяжелый проявления связаны с токсич.действием на клетки мозга высоких концентраций. Некоторые нарушения катаболизма тирозина в печени приводит к тирозинемии и тирозинурии. Причиной заболевания является дефект фетмента фумарилацетоацетатгидролазы. Клинические проявления-диарея,рвота. Тирозинемии 3 типов. 1 типтирозиноз – дефект фермента фумирилацетоацетатгидролазы, диарея,рвота,задержка в развитии; 2 тип синдром Рихнера – Ханхорта, дефект фермента тирозинаминтрансферазы, поражение глаз,кожи, умтс отсталость; новорожденных – снижение активности ферм п-гидроксифенилпируватдиоксигеназы. Энзимопатия. В основе многих заболеваний лежат нарушения функционирования ферментов в клетке-энзомопатии. При первичных энзимопатиях дефектные ферменты наследуются по аутосомно-рецессивному типу. Гетерозиготы не имеют фенотипических отклонений. Первичные энзимопатии обычно относят к метаболическим болезням, т.к. происходит нарушение определённых метаболических путей. Известно заболевание алкаптонурия. У таких больных наблюдают недостаточность фермента окисления гомогентизиновой кислоты. В присутствии кислорода эта кислота превращается в алкоптон. Алкаптон оседает в тканях,коже,суставах. Выделение с мочой темных пигментов алкаптонов. Нарушение образования конечных продуктов и накопление субстратов предшественников- это связано с нарушением распада гликогена в печени и выходом из неё глюкозы вследствие дефекта фермента 6-фосфатфосфатазы. Альбинизм – врожденный дефект тирозиназы, нарушается синтез пигментов меланинов. Болезнь Паркинсона - снижена активность тирозингидроксилазы, ДОФА-декарбоксилазы. Скованность, ригидность, тремор. 2. При стимуляции преганлионарного нейрона хромаффинные кл продуцируют катехоламины – дофамин, адреналин, норадреналин. Синтез происходит в цитоплазме и гранулах клеток мозгового слоя надпочечников. Катехоламины поступают в гранулы потеем АТФ-зависимого транспорта и хранятся в них. Освободившись путем экзоцитоза, В плазме крови катехоламины с альбумином образуют комплекс с альбумином. Адреналин транспортируется к печени и скелетным мышцам. Норадреналин в органах, иннервируемых симпатическими нервами. |