Реферат На тему адсорбция Студент Преподаватель Екатеринбург 2006г
 Скачать 1.06 Mb. Скачать 1.06 Mb.
|

1 2 Министерство образования уральская Государственная Сельскохозяйственная Академия. Кафедра химии Реферат На тему: адсорбция Студент__________________ Преподаватель__________________ Екатеринбург 2006г. Содержание. Адсорбция…………………………………………………………………………3 Абсорбция…………………………………………………………………………6 Сорбция...………………………………………………………………………….6 Поверхностное натяжение………………………………………………………..7 Хемосорбция………………………………………………………………………8 Смачивание………………………………………………………………………..8 Капиллярная конденсация………………………………………………………..9 Дисперсные системы…………………………………………………………….10 Мицелла…………………………………………………………………………..11 Коагуляция……………...………………………………………………………..12 Топохимические реакции………………………………………...……………..13 Катализ……...……………………………………………………………………14 Список литературы………………………………………………………………19 Адсорбция (от лат. ad — на, при и sorbeo — поглощаю), поглощение к.-л. вещества из газообразной среды или раствора поверхностным слоем жидкости или твёрдого тела. Например, если поместить в водный раствор уксусной кислоты кусочек угля, то произойдёт А. — количество кислоты в растворе уменьшится, молекулы кислоты сконцентрируются на поверхности угля. А. и абсорбция — поглощение в объёме тела, объединяются общим термином сорбция. Явление А. стало изучаться со 2-й половины 18 в. (Шееле, 1773), хотя несомненно, что в практической деятельности человечества А. использовалась с незапамятных времён. Учение об А. является частью более общей теории многокомпонентных гетерогенных систем, основы которой заложены У. Гиббсом (1876). Явление А. тесно связано с особыми свойствами вещества в поверхностном слое. например, молекулы, лежащие на поверхности раздела фаз жидкость — пар, втягиваются внутрь жидкости, т. к. испытывают большее притяжение со стороны молекул, находящихся в объёме жидкости, чем со стороны молекул пара, концентрация которых во много раз меньше концентрации жидкости. Это внутреннее притяжение заставляет поверхность сокращаться и количественно характеризуется поверхностным натяжением. По той же причине молекулы какого-либо другого вещества, оказавшиеся вблизи поверхности, притянутся к ней и произойдёт А. После А. внутреннее притяжение частично компенсируется притяжением со стороны адсорбционного слоя и поверхностное натяжение уменьшается. Гиббс вывел формулу, связывающую значение А. с изменением поверхностного натяжения. Те вещества, А. которых сильно уменьшает поверхностное натяжение, принято называть поверхностно-активными. Вещество, на поверхности которого происходит А., называется адсорбентом, а поглощаемое из объёмной фазы — адсорбатом. В зависимости от характера взаимодействия между молекулой адсорбата и адсорбентом А. принято подразделять на физическую А. и хемосорбцию. Менее прочная физическая А. не сопровождается существенными изменениями молекул адсорбата. Она обусловлена силами межмолекулярного взаимодействия, которые связывают молекулы в жидкостях и некоторых кристаллах и проявляются в поведении сильно сжатых газов. При хемосорбции молекулы адсорбата и адсорбента образуют химические соединения. Часто А. обусловлена и физическими и химическими силами, поэтому не существует чёткой границы между физикой А. и хемосорбцией. Физически адсорбированные молекулы более или менее свободно перемещаются по поверхности, при этом их свойства часто аналогичны свойствам очень тонкого слоя газа, т. н. двухмерного газа. Они могут собираться группами, образуя слой двухмерной жидкости или двухмерного твёрдого тела. Адсорбированные молекулы рано или поздно покидают поверхность — десорбируются. Время, в течение которого молекула находится на поверхности, называется временем А. Времена А. могут колебаться в очень широких пределах. Скоростью А. (соответственно скоростью десорбции) называется количество молекул, адсорбирующихся (или десорбирующихся) за единицу времени, оба значения величин относят к единице поверхности или массы адсорбента. Скорость хемосорбции, как и скорость любого химического процесса, чаще всего увеличивается с повышением температуры (т. н. активированная А., см. Хемосорбция). Если скорости А. и десорбции равны друг другу, то говорят, что установилось адсорбционное равновесие. В состоянии равновесия количество адсорбированных молекул остаётся постоянным сколь угодно долго, если неизменны внешние условия (давление, температура и др.). Адсорбированные молекулы не только совершают движение вдоль поверхности адсорбента, но и колеблются, то приближаясь к поверхности, то удаляясь от неё. Чем выше температура, тем интенсивнее колебательное движение, а стало быть, больше вероятность того, что в процессе таких колебаний связь молекулы с поверхностью будет разорвана и молекула десорбируется. Благодаря этому с ростом температуры уменьшается время А. и равновесное количество адсорбированных молекул. С ростом концентрации или давления адсорбата в объёме увеличивается частота попаданий молекул адсорбата на поверхность адсорбента; пропорционально ей возрастает скорость А. и увеличивается равновесное количество адсорбированных молекул. Кривые зависимости равновесной А. от концентрации или давления адсорбата при постоянной температуре называются изотермами А. Если адсорбат покрывает поверхность слоем толщиной в одну молекулу, А. называется мономолекулярной. Простейшая изотерма мономолекулярной А. представляет собой прямую линию, выходящую из начала координат, где на оси абсцисс отложено давление адсорбата Р, а на оси ординат степень заполнения поверхности Q, т. е. доля поверхности, покрытая адсорбированными молекулами. Это — т. н. изотерма Генри: Q = kP. Коэффициент пропорциональности k зависит главным образом от температуры и характера взаимодействия адсорбент — адсорбат. Уравнение Генри справедливо при очень низких степенях заполнения для однородной поверхности. По мере увеличения степени заполнения всё большую роль начинает играть взаимодействие между адсорбированными молекулами и интенсивность их поверхностной подвижности. Если молекулы адсорбата притягиваются друг к другу, то каждая вновь адсорбирующаяся молекула будет испытывать притяжение и адсорбата и молекул, адсорбированных ранее. Поэтому, по мере заполнения поверхности, силы, удерживающие адсорбированную молекулу, будут увеличиваться и условия для А. будут всё более и более благоприятными. В этом случае с ростом давления изотерма всё круче и круче идёт вверх (см. кривую 1). Однако по мере заполнения поверхности вновь адсорбирующимися молекулами становится всё труднее найти свободное (не занятое др. молекулами адсорбата) место на поверхности. Поэтому с увеличением давления рост А. замедляется и степень покрытия стремится к постоянному значению, равному единице (см. кривую 2, которая характерна при отсутствии взаимного притяжения молекул адсорбата). Если действуют оба эти фактора, то получаются вогнуто-выпуклые изотермы (см. кривую 3). Выпуклые изотермы (см. кривую 2) часто описывают уравнением Ленгмюра 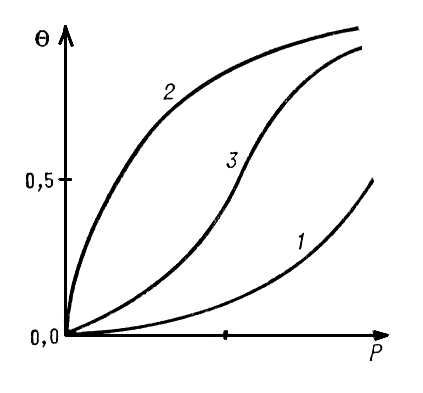 Здесь а — адсорбционный коэффициент, аналогичный по физическому смыслу константе Генри k. Уравнение Ленгмюра справедливо для мономолекулярной А. на однородной поверхности, если можно пренебречь притяжением молекул адсорбата между собой и их подвижностью вдоль поверхности. При дальнейшем увеличении давления происходит заполнение второго, третьего и т. д. слоев, т. е. имеет место полимолекулярная А. Если адсорбент имеет узкие поры и смачивается адсорбатом (см. Смачивание), то в порах может произойти конденсация при давлениях более низких, чем давление насыщенного пара адсорбата. Это явление называется капиллярной конденсацией. Поверхность твёрдых адсорбентов чаще всего неоднородна по адсорбционным свойствам: одни участки поверхности адсорбируют лучше, другие — хуже. При малых давлениях преобладает А. на наиболее активных участках поверхности, с увеличением давления заполняются менее активные участки. Однако, строго говоря, А. происходит одновременно на всей поверхности, и получаемая на опыте изотерма представляет собой сумму изотерм, каждая из которых соответствует определённому типу поверхности. Благодаря этому экспериментальные изотермы мономолекулярной А. могут существенно отличаться от кривых, приведённых на рис. Почти всегда процесс А. сопровождается выделением тепла, называемой теплотой А. Хотя теплота А. не является единственным фактором, характеризующим прочность А., однако чаще всего чем прочнее А., тем больше её теплота. Теплота хемосорбции обычно составляет несколько десятков ккал/моль, теплота физической А. редко превосходит 10 ккал/моль (40 кдж/моль). По мере заполнения неоднородной поверхности теплота А. обычно уменьшается. При переходе в область полимолекулярной А. теплота А. понижается до величины, близкой к теплоте конденсации адсорбата. А. играет важную роль при теплообмене между газообразными, жидкими и твёрдыми телами. например, молекулы газа, адсорбируясь на горячей поверхности, приобретают энергию, соответствующую температуре поверхности, и после десорбции сообщают эту энергию другим молекулам газа, нагревая его. Это не единственный, но важный механизм теплообмена. А.— один из решающих факторов в стабилизации коллоидных систем (см. Дисперсные системы, Мицелла, Коагуляция) и одна из важнейших стадий реакций в гетерогенных системах, в частности в гетерогенном катализе (см. Топохимические реакции, Катализ). В биологических системах А. — первая стадия поглощения субмикроскопическими коллоидными структурами, органеллами, клетками и тканями различных веществ из окружающей среды, функционирование биологических мембран, первые этапы взаимодействия ферментов с субстратом, защитные реакции против токсичных веществ, процессы всасывания — всё это связано с А. Многие адсорбенты (активный уголь, каолин, иониты и др.) служат противоядиями, поглощая и удаляя из организма попавшие в желудочно-кишечный тракт вредные вещества. А. применяется для разделения газовых и жидких смесей, для осушки и очистки газов и жидкостей (например, очистки воздуха в противогазах). Одним из древнейших применений А. является очистка вина. В науке и технике приобрёл большое значение хроматографический метод анализа, основанный на различной способности компонентов анализируемой смеси к А. (см. Хроматография). А. используют также для получения и очистки биологически активных веществ — витаминов, ферментов, гормонов, антибиотиков и др. При крашении тканей, в полиграфической промышленности имеют дело с А. молекул красителей. При производстве полимеров наполнителями служат адсорбенты. В вакуумной технике А. на стенках откачиваемой аппаратуры замедляет скорость откачки и ухудшает вакуум, однако, с другой стороны, действие различных сорбционных насосов основано на явлении А. В радиоэлектронной промышленности А. используется для стабилизации электрических свойств полупроводниковых приборов. Вообще во всех явлениях и процессах, где существенны поверхностные свойства, А. играет важную роль. Абсорбция (лат. absorptio — поглощение, от absorbeo — поглощаю), поглощение веществ из газовой смеси жидкостями. В технике А. обычно пользуются для извлечения из газовой смеси какого-либо компонента. Поглощение, точнее извлечение из жидкости какого-либо компонента жидкостью ранее также называлось А.; ныне такой процесс именуют экстракцией. При А. абсорбент поглощает всем своим объёмом. Скорость А. зависит от того, насколько концентрация поглощаемого газа в газовой смеси превосходит концентрацию этого компонента над раствором. Если концентрация растворяемого компонента в газовой смеси меньше его концентрации над жидкостью, растворяемый компонент выделяется из раствора (см. Десорбция). А. часто сопровождается химическим взаимодействием поглощаемого вещества с поглотителем (см. Хемосорбция). А. улучшается с повышением давления и понижением температуры. На А. основаны многие важнейшие промышленные процессы, например производство азотной, соляной и серной кислот (поглощение водой газообразных двуокиси азота, хлористого водорода и серного ангидрида), производство соды (А. углекислого газа), очистка отходящих промышленных газов от вредных примесей (сероводорода, сернистого ангидрида, окиси углерода, углекислого газа и др.), извлечение углеводородных газов и примесей (например, т. н. газового бензина, газов крекинга и пиролиза), а также выделение индивидуальных углеводородов. А. осуществляют на абсорбционных установках, основным аппаратом в которых служит абсорбер. Сорбция (от лат. sorbeo — поглощаю), поглощение твёрдым телом или жидкостью вещества из окружающей среды. Поглощающее тело называется сорбентом, поглощаемое им вещество — сорбатом (или сорбтивом). Различают поглощение вещества всей массой жидкого сорбента (абсорбция); поверхностным слоем твёрдого или жидкого сорбента (адсорбция). Поглощение вещества из газовой среды всей массой твёрдого тела или расплава называется такжеокклюзией. С., сопровождающаяся химическим взаимодействием сорбента с поглощаемым веществом, называется хемосорбцией. При С. паров высокопористыми телами часто имеет место капиллярная конденсация. В сорбционных процессах различные виды С. обычно протекают одновременно. (О применении С. см. Поверхностные явления, Ионный обмен, Хроматография.) В биологических системах большую роль играет С. (адсорбция) определённых веществ на поверхности клеток и мембранах внутриклеточных структур, а также С. (абсорбция) органоидами клетки и молекулами биополимеров. Для биологических систем характерна высокая специфичность (избирательность) С., что определяется особенностями пространственной конфигурации молекул сорбента. Эти макромолекулы играют роль рецепторов для соответствующего сорбата. Примерами С. может служить связывание молекул CO2 хлоропластами при фотосинтезе у растений, аминокислот — эритроцитами, переносящими их к тканевым клеткам, прикрепление фага к поверхности чувствительных к нему бактериальных клеток и др. Поверхностное натяжение, важнейшая термодинамическая характеристика поверхности раздела фаз (тел), определяемая как работа обратимого изотермического образования единицы площади этой поверхности. В случае жидкой поверхности раздела П. н. правомерно также рассматривать как силу, действующую на единицу длины контура поверхности и стремящуюся сократить поверхность до минимума при заданных объёмах фаз. Применительно к легкоподвижным поверхностям оба определения равнозначны, но первое предпочтительнее, т.к. имеет более ясный физический смысл. П. н. на границе двух конденсированных фаз обычно называется межфазным натяжением. Работа образования новой поверхности затрачивается на преодоление сил межмолекулярного сцепления (когезии) при переходе молекул вещества из объёма тела в поверхностный слой. Равнодействующая межмолекулярных сил в поверхностном слое не равна нулю (как в объёме тела) и направлена внутрь фазы с большей когезией. Таким образом, П. н. — мера некомпенсированности межмолекулярных сил в поверхностном (межфазном) слое или, что то же, избытка свободной энергии в поверхностном слое по сравнению со свободной энергией в объёмах соприкасающихся фаз. В соответствии с определениями П. н. его выражают в дж/м2 или н/м (эрг/см2 или дин/см). Благодаря П. н. жидкость при отсутствии внешних силовых воздействий принимает форму шара, отвечающую минимальной величине поверхности и, следовательно, наименьшему значению свободной поверхностной энергии. П. н. не зависит от величины и формы поверхности, если объёмы фаз достаточно велики по сравнению с размерами молекул; при повышении температуры, а также под действием поверхностно-активных веществ оно уменьшается. Расплавы металлов имеют наибольшее среди жидкостей П. н., например у платины при 2000 °С оно равно 1820 дин/см, у ртути при 20 °С — 484. П. н. расплавленных солей значительно меньше — от нескольких десятков до 200—300. П. н. воды при 20 °С — 72,8, а большинства органических растворителей — в пределах 20—60. Самое низкое при комнатной температуре П. н. — ниже 10 — имеют некоторые фторуглеродные жидкости. В общем случае многокомпонентных систем в соответствии с термодинамическим уравнением Гиббса при адсорбции изменение П. н. — ds = Г1dm1 + Г2dm2 +..., где Г1, Г2,... — поверхностные избытки компонентов 1, 2,..., т. е. разность их концентраций в поверхностном слое и объёме раствора (или газа), a dm1, dm2,... —изменения химических потенциалов соответствующих компонентов (знак «минус» показывает, что П. н. при положительной адсорбции уменьшается). Разницей в П. н. чистой жидкости и жидкости, покрытой адсорбционным монослоем, определяется поверхностное давление. На легкоподвижных границах жидкость — газ (пар) или жидкость — жидкость П. н. можно непосредственно измерить многими методами. Так, широко распространены способы определения П. н. по массе капли, отрывающейся от конца вертикальной трубки (сталагмометра); по величине максимального давления, необходимого для продавливания в жидкость пузырька газа; по форме капли (или пузырька), лежащей на плоской поверхности, и т.д. Экспериментальное определение П. н. твёрдых тел затруднено из-за того, что их молекулы (или атомы) лишены возможности свободного перемещения. Исключение составляет пластическое течение металлов при температурах, близких к точке плавления. Ввиду анизотропии кристаллов П. н. на разных гранях кристалла различно. Понятия П. н. и свободной поверхностной энергии для твёрдых тел не тождественны. Дефекты кристаллической решётки, главным образом дислокации, ребра и вершины кристаллов, границы зёрен поликристаллических тел, выходящие на поверхность, вносят свой вклад в свободную поверхностную энергию. П. н. твёрдых тел обычно определяют косвенно, исходя из межмолекулярных и межатомных взаимодействий. Величиной и изменениями П. н. обусловлены многие поверхностные явления, особенно в дисперсных системах (см. также Капиллярные явления), Л. А. Шиц. Хемосорбция, химическая сорбция, поглощение жидкостью или твёрдым телом веществ из окружающей среды, сопровождающееся образованием химических соединений. В более узком смысле Х. рассматривают как химическое поглощение вещества поверхностью твёрдого тела, т. е. как химическую адсорбцию. При Х. выделяется значительное количество тепла: обычно теплоты Х. лежат в пределах 84—126 кдж/моль (20—30 ккал/моль), а в некоторых случаях, например при Х. кислорода на металлах, могут превышать 420 кдж/моль (100 ккал/моль). Подобно химическим реакциям, Х. требует, как правило, значительной энергии активации. Поэтому при повышении температуры Х. ускоряется (т. н. активированная адсорбция). Х. избирательна, т. е. зависит от химического сродства адсорбируемого вещества к поверхности твёрдого тела. Для изучения Х. применяют физические методы: спектроскопию, электронный парамагнитный и ядерный магнитный резонанс, электронный и ионный проекторы, дифракцию медленных электронов и др. Х. играет большую роль в гетерогенном катализе, очистке газов, вакуумной технике и др Смачивание, явление, возникающее при соприкосновении жидкости с поверхностью твёрдого тела или другие жидкости. Оно выражается, в частности, в растекании жидкости по твёрдой поверхности, находящейся в контакте с газом (паром) или другой жидкостью, пропитывании пористых тел и порошков, искривлении поверхности жидкости у поверхности твёрдого тела. Так, С. вызывает образование сферического мениска в капиллярной трубке, определяет форму капли на твёрдой поверхности или форму газового пузырька, прилипшего к поверхности погруженного в жидкость тела. С. часто рассматривают как результат межмолекулярного (вандерваальсова) взаимодействия в зоне контакта трёх фаз (тел, сред). Однако во многих случаях, например при соприкосновении жидких металлов с твёрдыми металлами, окислами, алмазом, графитом, С. обусловлено не столько межмолекулярным взаимодействием, сколько образованием химических соединений, твёрдых и жидких растворов, диффузионными процессами в поверхностном слое смачиваемого тела. Тепловой эффект, сопровождающий соприкосновение жидкости со смачиваемой поверхностью, называется теплотой смачивания. Мерой С. обычно служит краевой угол q между смачиваемой поверхностью и поверхностью жидкости на периметре С. (рис. 1). Угол q отсчитывают со стороны жидкости. При статическом (равновесном) С. он связан с поверхностным натяжением жидкости (sж), поверхностным натяжением твёрдого тела (sт) и межфазным натяжением на границе твёрдое тело — жидкость (sтж) уравнением Юнга: cosq = (sт — sтж)/(ж. Величиной угла q оценивают лиофильностьилиофобность поверхностей по отношению к различным жидкостям. На лиофильной поверхности жидкость растекается, т. е. имеет место частичное (0° С. имеет важное значение в природе, промышленной технологии, быту. Хорошее С. необходимо при крашении и стирке (см. Моющее действие), обработке фотографических материалов, нанесении лакокрасочных покрытий, пропитке волокнистых материалов, склеивании, пайке, амальгамировании и т. д. Снизить С. до минимума стремятся при получении гидрофобных покрытий, гидроизоляционных материалов и др. В некоторых случаях, например при флотации и эмульгировании твёрдыми эмульгаторами, требуется сохранение краевых углов в определённом интервале значений. С. играет первостепенную роль в металлургических процессах, при диспергировании твёрдых тел в жидкой среде. Оно влияет на распространение грунтовых вод, увлажнение почв, разнообразные биологические и другие природные процессы. В развитие теории и разработку прикладных вопросов С. большой вклад внесли П. А. Ребиндер, А. Н. Фрумкин, Б. В. Дерягин и др Капиллярная конденсация, конденсация пара в капиллярах и микротрещинах пористых тел или в промежутках между тесно сближенными твёрдыми частицами. Необходимым условием К. к. является смачивание жидкостью поверхности тела (частиц). К. к. начинается с адсорбции молекул пара поверхностью конденсации и образования менисков жидкости. При вогнутой форме менисков давление насыщенного пара над ними, согласно Кельвина уравнению, ниже, чем давление насыщенного пара po над плоской поверхностью. В результате К. к. происходит при более низких давлениях пара, чем давление насыщения po. Объём сконденсировавшейся в порах жидкости достигает предельной величины при внешнем давлении пара р = ро. В этом случае поверхность раздела жидкость — газ имеет нулевую кривизну (плоскость, катеноид), Сложная капиллярная структура пористого тела может служить причиной капиллярного гистерезиса — зависимости количества сконденсировавшейся в порах жидкости не только от давления пара, но и от предыстории процесса, т. е. от того, как было достигнуто данное состояние: в процессе конденсации или же в ходе испарения жидкости, К. к. увеличивает поглощение (сорбцию) паров пористыми телами, в особенности вблизи точки насыщения паров. К. к. используется в промышленности для улавливания жидкостей тонкопористыми телами (сорбентами). Большую роль К. к. играет также в процессах сушки, удержания влаги почвами, строительными и др. пористыми материалами (см. Капиллярные явления). Дисперсные системы, образования из двух или большего числа фаз (тел) с сильно развитой поверхностью раздела между ними. В Д. с. по крайней мере одна из фаз — дисперсная фаза — распределена в виде мелких частиц (кристалликов, нитей, плёнок или пластинок, капель, пузырьков) в другой, сплошной, фазе — дисперсионной среде. Д. с. по основной характеристике — размерам частиц или (что то же самое) дисперсности (определяемой отношением общей площади межфазной поверхности к объёму дисперсной фазы) — делятся на грубо (низко) дисперсные и тонко (высоко) дисперсные, или коллоидные системы (коллоиды). В грубодисперсных системах частицы имеют размер от 10-4 см и выше, в коллоидных — от 10-4—10-5 до 10-7 см. По агрегатному состоянию дисперсионной среды различают газодисперсные системы — аэрозоли (туманы, дымы), пыль; жидкодисперсные — золи, суспензии, эмульсии, пены; твёрдодисперсные — стеклообразные или кристаллические тела с включениями мельчайших твёрдых частиц, капель жидкости или пузырьков газа (см. табл.). Пыль, суспензии, лиофобные эмульсии (см. Лиофильные и лиофобные коллоиды) — грубодисперсные системы; как правило (при наличии разности плотностей), они седиментационно неустойчивы, т. е. их частицы оседают под действием силы тяжести или всплывают. Золи — типичные высокодисперсные коллоидные системы, частицы дисперсной фазы которых (мицеллы) участвуют в броуновском движении и потому седиментационно устойчивы. Жидкие и твёрдые пены, состоящие из газовых ячеек-пузырьков, разделённых тонкими прослойками непрерывной фазы, представляют особую группу структурированных ячеистых систем (см. ниже). По интенсивности молекулярного взаимодействия фаз различают лиофильные и лиофобные Д. с. В лиофильных системах молекулярное взаимодействие между фазами достаточно велико и удельная свободная поверхностная энергия (поверхностное натяжение) на межфазной границе очень мала. Лиофильные системы образуются самопроизвольно (спонтанно) и имеют предельно высокую дисперсность. В лиофобных системах взаимодействие между молекулами различных фаз значительно слабее, чем в случае лиофильных систем; межфазное поверхностное натяжение велико, вследствие чего система проявляет тенденцию к самопроизвольному укрупнению частиц дисперсной фазы (см. Коагуляция и Коалесценция). Обязательное условие существования лиофобных Д. с. — наличие стабилизаторов, веществ, которые адсорбируются на поверхности раздела фаз и образуют защитные слои, препятствующие сближению частиц дисперсной фазы. Д. с. могут быть бесструктурными (свободнодисперсными) и структурированными (связнодисперсными). Структурированные Д. с. пронизаны сеткой-каркасом из соединённых между собой частиц (капель, пузырьков) дисперсной фазы, вследствие чего обладают некоторыми механическими свойствами твёрдых тел (подробнее см. Дисперсная структура, Гели). Характерная особенность Д. с. — высокая свободная энергия как следствие сильно развитой межфазной поверхности; поэтому Д. с. обычно (кроме лиофильных Д. с.) термодинамически неустойчивы. Они обладают повышенной адсорбционной способностью, химической, а иногда и биологической активностью. Д. с. — основной объект изучения коллоидной химии. Д. с. широко распространены в природе, технике и быту. Примерами Д. с. могут служить горные породы, грунты, почвы, дымы, облака, атмосферные осадки, растительные и животные ткани; строительные материалы, краски, моющие средства, волокнистые изделия, важнейшие пищевые продукты и многие др. Классификация дисперсных систем по агрегатному состоянию фаз
1 2 | |||||||||||||||||||||