Курсовая работа Трошин. Российский университет дружбы народов
 Скачать 97.55 Kb. Скачать 97.55 Kb.
|

|
Федеральное государственное автономное образовательное учреждение высшего образования "РОССИЙСКИЙ УНИВЕРСИТЕТ ДРУЖБЫ НАРОДОВ" (РУДН) Институт биохимической технологии и нанотехнологии Промышленная фармация Курсовая работа «Валидация фармацевтического производства. Валидация стерилизующей фильтрации. Исполнитель: Трошин Алексей Павлович ЦПФмз-01-20 Москва 2021 СодержаниеВведение 3 1. Основные понятия 6 Валидация в Российской федерации 8 Квалификация 11 Валидация 14 Валидация стерилизующей фильтрации 17 Использование механических фильтров в производстве 22 Заключение 27 Используемая литература 29 ВведениеВ настоящее время в практике одним из важнейших документов, требования к производству и контролю качества ЛЕКАРСТВЕННЫЕ СРЕДСТВА для человека и животных, являются "Правила производства ЛЕКАРСТВЕННЫЕ СРЕДСТВА " - "Good Manufacturing Practice for Medicinal Products (GMP)". Это означает, что условием обеспечения качества лекарств является их производство в соответствии с правилами надлежащей практике . В основных требованиях GMP, предъявляемых к производству ЛЕКАРСТВЕННЫЕ СРЕДСТВА , является предотвращение их перекрестного загрязнения. ЛЕКАРСТВЕННЫЕ СРЕДСТВА могут быть загрязнены другими препаратами или активными фармацевтическими субстанциями, моющими или дезинфицирующими средствами, микроорганизмами, частицами пыли, смазочными материалами, вспомогательными веществами, промежуточной продукцией и др. В тоже время во многих случаях при производстве различных препаратов используется одно и то же оборудование. Поэтому для предотвращения загрязнения каждого последующего препарата предыдущим или предыдущей серией того же наименования очень важным является проведение эффективной процедуры очистки оборудования, с обязательной оценкой степени его чистоты и валидации этого процесса. Не менее важным является контроль качества исходных веществ или субстанций, в том числе и их микробиологическая чистота. Особенно важен этот аспект при производстве асептических лекарственных средств и лекарственных препаратов, предназначенных для детей. Для обеспечения постоянства качества при производстве лекарств установлена обязательность проведения валидации на фармацевтическом производстве в разделе "Валидация" ОСТ 42-510-98 "Правила производства и контроля качества лекарственных средств GMP", а также в ГОСТ Р 52249-2009 "Правила производства и контроля качества лекарственных средств". Основные задачи российской фармацевтической промышленности можно сформулировать как обеспечение населения безопасными и эффективными лекарственными средствами, гармонизация с международными требованиями российских стандартов по разработке и производству лекарственных средств, стимулирование разработки и производства инновационных лекарственных средств, поддержка экспорта российских лекарств, защита внутреннего рынка от недобросовестной конкуренции и выравнивание условий доступа на рынок для отечественных и зарубежных производителей, осуществление технологического перевооружения российской фармацевтической отрасли. В соответствии с поставленными задачами проводится ряд мероприятий в фармацевтической отрасли России. В России Правила производства лекарственных средств (Good Manufacturing Practice for Medicinal Products, GMP) начали внедряться с 2004 года, после принятия ГОСТ Р 522549-2004 "Правила производства и контроля качества лекарственных средств". Сегодня действует обновленная редакция ГОСТ Р 52249-2009. Руководство НАДЛЕЖАЙЩАЯ ПРОИЗВОДСТВЕННАЯ ПРАКТИКА применяется для построения систем обеспечения качества и организации надлежащего производства готовых лекарственных средств и действующих веществ – для проектирования, строительства, реконструкции и технического переоснащения предприятий фармацевтического профиля, для аудита, инспектирования и сертификации производственных участков. В соответствие с практикой , одним из основных принципов построения системы обеспечения качества фармацевтического производства является проведение аттестации/квалификации (валидации) технологических процессов, оборудования, помещений, инженерных систем, методов анализа с целью подтверждения соответствия заранее установленным спецификациям и характеристикам и параметрам. В связи с тем, что валидация чрезвычайно важна для выпуска качественной фармацевтической продукции, мы рассмотрим основные вопросы, касающиеся этого аспекта фармацевтического производства. 1. Основные понятияВалидация (Validation) – документированная процедура, дающая высокую степень уверенности в том, что конкретный процесс, метод или система будет последовательно приводить к результатам, отвечающим заранее установленным критериям приемлемости. Валидационный план (Validation Master Plan) – документ, который описывает философию, стратегию и методологию предприятия по проведению валидации. Валидационный протокол – документ, отражающий результаты валидации процессов (PV) и квалификации: проектной документации (DQ), монтажа (IQ), функционирования (OQ) и эксплуатации (PQ) оборудования, инженерных систем, "чистых помещений" и др. Квалификация (Qualification) – оценка и документированное подтверждение того, что проектная документация, оборудование, инженерные системы и другие условия производства способны обеспечить достижение ожидаемых и воспроизводимых результатов. Квалификация проектной документации (Design Qualification – DQ) – оценка и документированное подтверждение соответствия проектной документации требованиям правил GMP. Квалификация монтажа (Installation Qualification – IQ) – оценка и документированное подтверждение соответствия качества монтажа/установки технологического и лабораторного оборудования, инженерных систем, "чистых" помещений и др., требованиям нормативной и технической документации. Квалификация функционирования (Operational Qualification – OQ) – оценка и документированное подтверждение соответствия работоспособности технологического и лабораторного оборудования, инженерных систем, оснащенных "чистых" помещений и др., требованиям нормативной и технической документации. Квалификация эксплуатации (Performance Qualification – PQ) – оценка и документированное подтверждение соответствия надежности и эффективности эксплуатационных параметров технологического оборудования, инженерных систем, функционирующих (чистых( помещений и др., требованиям нормативной и технической документации. ЛС – вещества, применяемые для профилактики, диагностики, лечения болезней, предотвращения беременности; полученные из крови, плазмы крови, а так же органов, тканей человека или животного, растений, микроорганизмов, минералов, методами синтеза или с применением биологических технологий. К лекарственным средствам относятся так же вещества растительного, животного или синтетического происхождения, обладающие фармакологической активностью и предназначенные для изготовления лекарственных препаратов. Отчет о проведении валидации – документ предприятия, отражающий и оценивающий результаты валидации процессов (PV) и всех стадий квалификации (DQ, IQ, OQ, PQ). Валидация в Российской федерацииВ России требования к валидации изложены в ряде нормативных документов, которые содержат общие подходы к процедурам, носят формальный или рекомендательный характер без конкретных методов и примеров. Кроме этого, проведение валидации должно сопровождаться грамотным документооборотом на предприятии, что также затруднительно без четких и единых рекомендаций со стороны разработчиков нормативных документов. В современном мире должно быть оформлено электронно и заархивировано Валидация – раздел правил GMP, касающийся надежности условий производства и их способности приводить к ожидаемым результатам по показателям качества продукции. Валидация является важной частью системы обеспечения и контроля качества. Валидация сама по себе не улучшает качества продукции. Ее результаты могут либо повысить степень гарантии качества, либо указать на необходимость совершенствования условий производства. И данный процесс может выполнить воиспроизводимость с заданными параметрами Только валидация может дать уверенность, что при данных условиях и с такими производственными параметрами фармацевтическое производство способно выпускать лекарственные препараты необходимого качества. В фармацевтической отрасли, термин "валидация" трактуется следующим образом: "Процесс документированного подтверждения достижения разумной степени уверенности в том, что Производственный процесс, Аналитические методики, спользуемое оборудование, Производственные системы, соответствуют действующим принципам GMP и выполняют свое функциональное назначение, т.е. их использование действительно дает ожидаемые результаты". По сути, валидация технологического процесса – это конечная цель, для достижения которой нужно последовательно провести валидацию ряда других связанных процессов. В GMP общий термин "валидация" разделяется на два понятия: "валидация процессов" и "квалификация производственных систем". Квалификация производственных систем – это часть валидации процесса, направленная на документальное подтверждение пригодности оборудования, инженерных систем, комплекса помещений, которые используются в производстве лекарственного препарата. Квалификация проводится для того, чтобы быть уверенными в том, что производственная система не влияет на качество продукта, а также для того, что если мы в ходе непосредственно валидации технологического процесса получим негативный результат – это не может быть связано с отказами оборудования/систем, а причины нужно искать в самом технологическом процессе). По своей логике, квалификация производственных систем – это некая предупреждающая мера. Таким образом, под валидацией процесса в фармацевтической отрасли подразумевается: Квалификация чистых помещений; Квалификация инженерных систем (подготовка чистого воздуха, воды очищенной и воды для инъекций, сжатого воздуха и т.п.); Квалификация производственного оборудования; Квалификация аналитического оборудования (используемого для контроля качества сырья, полупродуктов и готовой продукции); Квалификация складских зон (сырье, готовая продукция); Валидация компьютеризированных систем, включая квалификацию ИТ-инфраструктуры; Валидация аналитических методик; Верификация фармакопейных методик (методики, внесенные в национальную или региональные Фармакопеи); Валидация очистки помещений, оборудования; Валидация асептических условий; Валидация этапов технологического процесса; Валидация упаковки КвалификацияДля каждого критического объекта инфраструктуры должна быть проведена квалификация, которая, как правило, осуществляется в четыре последовательных этапа: Квалификация проекта (DQ, Design Qualification) Квалификация монтажа (IQ, Installation Qualification) Квалификация функционирования (OQ, Operational Qualification) Квалификация в эксплуатации (PQ, Performance Qualification) Квалификация проекта (DQ) направлена на документированное подтверждение пригодности проекта (конструкции, проектного решения) технических средств, инженерных систем и оборудования для их предполагаемого использования. Объем работ на этом этапе: Описание системы (функция, параметры оборудования, особые характеристики) Техническая документация (нормативные требования, документация по оборудованию) Оценка конструкции (конструкционные материалы, оценка риска загрязнений) Компоненты/элементы оборудования/системы Анализ возможных отказов/дефектов Анализ способа изготовления (критические параметры работ при изготовлении оборудования, требования по калибровке) Квалификация монтажа (IQ) направлена на документированное подтверждение того, что технические средства, инженерные системы и оборудование сконструированы, оснащены и смонтированы в соответствии с рабочей документацией проекта и рекомендациями производителя. Объем работ на этом этапе: Наличие достаточной документации Наличие всех элементов в поставке Правильность монтажа и подключений Соответствие контактирующих материалов Соответствие средств измерений Квалификация функционирования (OQ) направлена на документированное подтверждение того, что технические средства, инженерные системы и оборудование функционируют должным образом по всему заявленному диапазону рабочих характеристик. Объем работ на этом этапе: Приемлемость документации (инструкции по эксплуатации, обслуживанию); Испытания, включающие условие или ряд условий, охватывающих верхний и нижний пределы рабочих параметров: Срабатывание блокировок/сигнализаций. Как правило, после этого этапа квалификации объект вводится в эксплуатацию. Квалификация функционирования (PQ) проводится для инженерных систем, которые работают непрерывно, а также для оборудования со сложным управлением. Квалификация в эксплуатации – это документированное подтверждение того, что технические средства, инженерные системы и оборудование при совместном (или длительном) использовании могут надежно функционировать с получением воспроизводимых свойств продукта. При этом, если производственная система оснащена автоматизированной системой мониторинга параметров, или обработки данных, дополнительно должна проводиться валидация компьютеризированной системы. ВалидацияВалидация аналитических методик Каждая аналитическая и микробиологическая методика, которая используется для контроля, должна пройти валидацию. Это означает, что мы обязаны получить доказательства пригодности такой методики для контроля , гарантии получения достоверных результатов. В этом плане, требования GMP полностью совпадают с требованиями ИСО 17025. Валидация очистки Процедуры очистки оборудования должны также пройти валидацию до того, как мы приступим к производству препарата на этом оборудовании. Прежде всего, эта валидация направлена на получение гарантий возможности проведения качественной очистки после изготовления такого продукта. По сути, это минимизация риска перекрестного загрязнения при переходе на производство другого продукта на этом же оборудовании. Если на оборудовании останутся остатки предыдущего продукта, это не будет обнаружено – так как отсутствует аналитический контроль именно на наличие таких примесей. Валидация асептических условий При производстве стерильных лекарственных средств с использованием асептических технологий до начала самого технологического процесса необходимо подтвердить, что на всем протяжении процесса изготовления препарата (т.е. длительность процесса), в продукт не попадает ни один микроорганизм. Валидация асептических условий проводится по сценарию имитации с помощью питательных сред. Валидация технологического процесса И непосредственно, валидация каждого из этапов технологического процесса проводится на 3-х последовательных сериях с учетом "наихудшего случая". И, что очень важно, валидация технологического процесса проводится отдельно для каждого продукта и его заявляемого размера серии. (рис. 1). Наихудший случай – это проведение процесса при таких условиях и обстоятельствах (для параметров процесса, режимов работы оборудования), которые имеют максимальные шансы вызвать отклонение процесса или несоответствие продукта по сравнению с идеальными условиями. Логика очень проста – если при таких условиях мы получаем качественный продукт, значит, гарантированно мы будем достигать качества внутри заданных диапазонов. Повторная валидация/квалификация Через заданные периоды эксплуатации (использования), каждый объект/процесс должны пройти повторную валидацию. Основная цель повторной валидации (ревалидации) – это получить подтверждения того, что объект/процесс продолжает находиться в валидном состоянии. Это полностью отражает логику GMP: "Для подтверждения качества продукта недостаточно провести валидацию в начале его жизненного цикла, необходимо обеспечить мониторинг и постоянное улучшение". 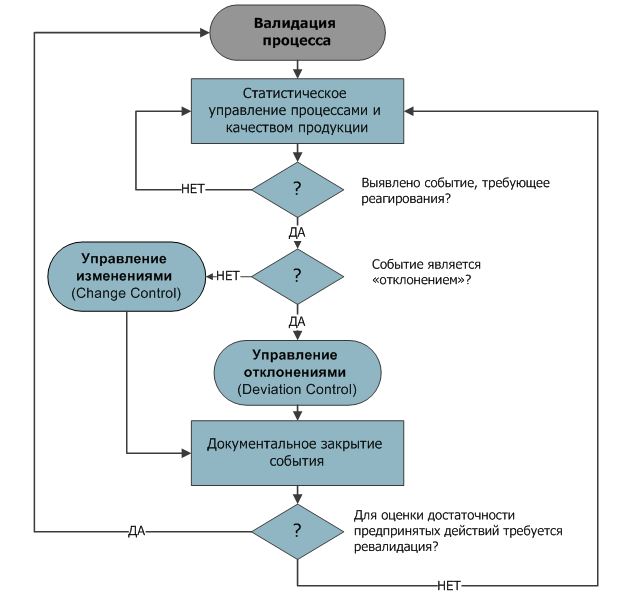 Рис. 1. Схема надзора над валидированным процессом. Ретроспективная валидация может проводиться только для хорошо отработанных процессов. Проведение ее не допускается, если в состав продукции, технологический процесс или оборудование недавно были внесены изменения. Источники данных для такой валидации должны включать (но не ограничиваться ими): протоколы производства и упаковки серий продукции, контрольные карты производства, журналы проведения технического обслуживания, данные об изменениях в персонале, исследования возможностей процесса, данные о готовой продукции, в том числе карты тенденций, а также результаты изучения ее стабильности при хранении. Перспективная валидация должна включать следующие элементы (но не ограничиваться ими): краткое описание процесса; перечень критических стадий процесса, подлежащих исследованию; перечень используемых помещений/оборудования (включая измерительное/контрольное/регистрирующее оборудование) с указанием сведений об их калибровке; спецификации на готовую продукцию при выпуске; при необходимости перечень аналитических методик; предлагаемые точки контроля в процессе производства и критерии приемлемости; при необходимости дополнительные испытания, которые следует провести, вместе с критериями приемлемости и валидацией аналитических методик; план отбора проб; методы регистрации и оценки результатов; функции и обязанности; предполагаемый график выполнения работ. С помощью установленного процесса (используя компоненты, соответствующие спецификациям) можно произвести ряд серий готовой продукции при обычных условиях. Теоретически количество выполненных производственных циклов и сделанных наблюдений должно быть достаточным, чтобы позволить установить обычную степень изменчивости и тенденции, а также получить необходимое количество данных для оценки. Для валидации процесса считается достаточным выполнить три последовательные серии/цикла, при которых параметры находятся в заданных пределах. При этом размер серии при валидации должен быть равным размеру серии при промышленном выпуске продукции. В исключительных случаях допускается начинать серийное производство до завершения программы валидации. фармацевтический лекарственный валидация инновационный Валидация стерилизующей фильтрацииВ настоящей пункте рассматриваются процедуры квалификации (валидации) мембранных фильтроэлементов патронного типа для стерилизующей фильтрации растворов, как основного элемента системы для проведения стерилизующей фильтрации, а именно на основе полиамида (Nylon6+66) марки ЭПМ.К-/020, ЭПМ.К+-/020 и полиэфирсульфона (PES) марки ЭПМ.ПС-/020, НА территории РФ в силу правил GMP, а именно документа «Правила организации производства и контроля качества лекарственных средств», разработанного под эгидой Минпромторга РФ, (далее – Правила), взамен действующего ГОСТ Р 52249-09 «Правила производства и контроля качества лекарственных средств» (далее – ГОСТ Р). И Правила, и ГОСТ Р предусматривают необходимость проведения валидации процессов получения лекарственных средств. В свою очередь для осуществления процедуры валидации необходима квалификация (аттестация) участвующего в техпроцессе оборудования. В соответствии с Правилами и ГОСТ Р (Часть 1, глава 3, п.3.34) «Конструкция, монтаж и порядок технического обслуживания производственного оборудования должны соответствовать его назначению». Кроме того, «Технологическое оборудование не должно влиять на качество продукции и представлять опасность для продукции. Части технологического оборудования, контактирующие с продукцией, не должны вступать с ней в химическую реакцию, выделять или абсорбировать вещества, оказывающие влияние на качество продукции». В соответствии с этими рекомендациями компаниями подготовлено «Руководство по валидации» мембранных фильтроэлементов для стерилизующей фильтрации жидкостей при их производстве, которое является материалом, необходимым для Потребителя для валидации всего технологического процесса стерилизации лекарственного средства методом фильтрования. Среди всех областей применения для очистки и разделения жидкостей и газов мембранные фильтры, используемые в производстве лекарственные средства, характеризуются самыми строгими требованиями к качеству и условиям их производства и применения, а фильтры для асептического производства растворов термолабильных веществ, где метод стерилизации является единственно возможным, наиболее жесткими. Такое повышенное внимание к процессу стерилизующего фильтрования объясняется еще и тем, что фильтрация и сегодня остается достаточно рискованным способом стерилизации. Гофрированные мембранные фильтроэлементы патронного типа, используемые в производстве лекарственных средств, характеризуются большой площадью поверхности мембранного полотна (до 1м2). Это обстоятельство предъявляет крайне высокие требования к однородности и стабильности исходного микропористого полотна, иначе при использовании невалидированных мембран, появляется риск нестерильности всего объема фильтрата лекарства, который может достичь десятка кубометров. Возможные дефекты фильтра при установке и сборке, нарушение целостности в процессе эксплуатации, когда подвергается гидравлическим и термическим нагрузкам в процессе фильтрации, тестирования, стерилизации автоклавированием и паром в линии - все эти риски диктуют жесткие требования к квалификации (валидации) ФЭ как при их производстве, так и при непосредственном использовании в процессах фильтрации ГЛС. Кроме того, возможность использования фильтров для медико-биологических задач определяется не только стерилизующей эффективностью, производительностью, механической прочностью, возможностью применения различных способов стерилизации, но и отсутствием выделения токсичных компонентов в стерилизуемую жидкость. Руководство по валидации включает: описание ФЭ и основные области его применения; обозначение ФЭ по каталогу; основные характеристики ФЭ; программу квалификационных испытаний; методы испытаний; результаты квалификационных испытаний образец сертификата качества Важно отметить, что все фильтрующие элементы производятся в условиях «чистого» помещения (классы чистоты 7 и 8 ИСО согласно ГОСТ Р ИСО 14644-5-2005, С и D согласно требований GMP на производство медицинской продукции) с соблюдением требований сертифицированной системы менеджмента качества согласно ГОСТ ISO 9001-2011 (ISO 9001:2008) и проходят контроль по специфическим показателям, указанным в программе квалификационных испытаний. В программу квалификационных испытаний стерилизующих мембранных фильтроэлементов входят: Испытания на целостность, включающие испытания на давление «точки пузырька» и скорость диффузии, корреляция данных неразрушающего метода контроля целостности с микробиологическим тестом. Измерение производительности по воде при разных перепадах давления. Определение сдвига рН при фильтрации. Тест на содержание частиц и волокон в фильтрате. Тестирование на биологическую безопасность. Определение содержания окисляемых веществ в фильтрате. Определение содержания экстрагируемых веществ в фильтрате. Определение содержания общего органического углерода в фильтрате. Определение содержания бактериальных эндотоксинов . Определение эффективности удержания и сорбционной емкости по бактериальным эндотоксинам. Определение стерилизующих свойств ФЭ по удержанию тест-культуры Brevundimonas diminuta. Определение термостойкости (стойкости к автоклавированию). Проверка биологической безопасности, установленная путем санитарно-химических и токсикологических испытаний. . Использование механических фильтров в производствеМеханические фильтры по очистке воды и воздуха используются во многих отраслях производства и промышленности. Наиболее значимой для повседневной жизни отрасли является медицина и фармакологическое производство. Неправильная фильтрация воздуха и воды на производстве может провести к несоблюдению исполнения технических условий и технического процесса, что в итоге отразится на качестве выпускаемой продукции, а значит и на потребителе, выбравшего данный товар для потребления. В основных требованиях надлежащей практике , предъявляемых к производству лекарствам , особое внимание уделяется предотвращению их от перекрестного загрязнения. При этом ответственным процессом является очистка технологического оборудования. Лекарственные препараты и активные фармацевтические субстанции могут быть загрязнены другими препаратами или активными фармацевтическими субстанциями, моющими или дезинфицирующими средствами, микроорганизмами, частицами пыли, смазочными материалами, вспомогательными веществами, промежуточной продукцией и др. Поэтому особенно важно максимально предотвратить опасность проникновения с воздухом или водой мелких и крупных частиц, способных загрязнить лекарственное средство[13]. Причинами попадания загрязняющих частиц в объекты производства с воздухом могут быть первичная высокая контаминация атмосферного воздуха, особенно если она попадает без предварительной очистки, и неэффективная работа системы воздухоподготовки. В зависимости от уровня требований, предъявляемых к микробной частоте воздуха, уже на стадии проектирования выбирают необходимое число степеней очистки. Нормы допустимого загрязнения воздуха в производственном помещении устанавливают в зависимости от характера технологического процесса и опасности загрязнения конечного продукта. Производственные помещения должны иметь эффективную систему приточной и вытяжной вентиляции с контролирующим воздушный поток оборудованием и приборами для измерения температуры, влажности, эффективности фильтрации и перепада давления на фильтрах. Чистым помещением называется помещение, в котором счётная концентрация аэрозольных частиц и, при необходимости, число микроорганизмов в воздухе поддерживаются в определённых пределах [4]. Класс чистого помещения характеризуется классификационным числом, определяющим максимально допустимую счётную концентрацию аэрозольных частиц определённого размера в 1 куб. метре воздуха. Следует различать три фазы создания и существования чистого помещения: построенное, когда чистое помещение построено и действует, но технологическое оборудование не установлено или не работает, а материалы и персонал отсутствуют; оснащенное, когда чистое помещение построено и действует, технологическое оборудование установлено и действует в соответствии с соглашением между покупателем и поставщиком, а персонал отсутствует; эксплуатируемое, когда чистое помещение функционирует в соответствии с предусмотренными требованиями и присутствует необходимый персонал На чистом участке или в чистом помещении с вертикальным ламинарным потоком фильтры приточной вентиляции должны располагаться в потолке, а отверстия вытяжной вентиляции - в полу или нижней части стен. В чистом помещении с горизонтальным ламинарным потоком фильтры приточной и отверстия вытяжной вентиляции должны располагаться на всей поверхности противоположных стен. Чистые камеры должны отвечать следующим требованиям: направляющие потоки панели, колпак и рабочие поверхности должны быть изготовлены из гладкого и прочного материала; фильтры предварительной очистки должны быть одноразовыми или из материала, позволяющего тщательно очищать их и использовать вновь; конечная фильтрация должна осуществляться через предварительно испытанные и герметично установленные фильтры тонкой очистки; скорость ламинарного потока должна быть в пределах 0,45 м/с +/- 20%. Необходимо регулярно проводить оценку эффективности работы воздушных фильтров с помощью контроля запыленности воздуха и DOP-теста (испытание на герметичность и утечку). Замена или герметизация фильтрующего оборудования должна проводиться строго по показаниям приборов в соответствии с нормативными требованиями. Сроки замены должны определяться при увеличении сопротивления потоку воздуха вдвое по сравнению с исходной нормативной величиной, что свидетельствует о снижении производительности фильтра или о возможности повреждения. Воду в производстве лекарственных веществ используют в качестве основного и вспомогательного материала. Вода является компонентом питательных сред и готовых лекарственных форм. Ее используют в технологии выделения и очистки биологически активных веществ, для санитарной подготовки помещений и оборудования, а также для приготовления растворов дезинфектантов и антисептиков. В технологических процессах используют питьевую воду из центральных систем хозяйственно-питьевого водоснабжения и очищенную воду, получаемую на производстве методами фильтрации. Вода очищенная используется для: изготовления инъекционных лекарственных средств; получения пара; санитарной обработки; мытья емкостей; в лабораторной практике. При этом механические фильтры используются для грубой и тонкой очитки воды, что является базовой стадией очистки воды на фармацевтическом производстве. Далее следует более углубленная очистка, обесцвечивание и обессоливание воды. Механические фильтры по очистке воды предотвращают от быстрой порчи более тонкие фильтры, такие как мембранные и катриджные и используются на стадии предварительной очистки воды. Технологическая схема предварительной очистки исходной воды в зависимости от источника водоснабжения и ее состава может включать в себя следующие стадии: стадию напорной аэрации (или безнапорной аэрации) воды; стадию пропорционального дозирования окислителя (биоцида) и коагулянта (флокулянта) с помощью насосов-дозаторов для разрушения ассоциатов и комплексов, а также для удаления как растворенного в воде диоксида кремния, так и коллоидных соединений кремния, железа и др.; стадию очистки на однослойных или многослойных насыпных фильтрах (фильтрах механической очистки, фильтрах обезжелезивания); при высоком содержании кремния в исходной воде следует отказываться от фильтрации с использованием кремнийсодержащих фильтрующих сред (например, от использования кварцевого песка); стадию адсорбционной очистки на насыпных адсорбционных фильтрах с активным углем, обладающим развитой структурой мезапор, для удаления свободного хлора, присутствующего в воде, органических соединений, влияющих на значение общего органического углерода; стадию тонкой очистки на мультипатронных фильтрах с использованием картриджей (фильтрующих элементов) глубинного типа; стадию ультрафиолетовой стерилизации (УФ-стерилизации) с длиной волны 254 нм для предотвращения микробиологического загрязнения стадии обессоливания воды; стадию ультрафильтрации. При необходимости в систему предварительной очистки воды включаются: стадия умягчения воды (Na-катионирования), необходимая для удаления из воды солей жесткости и обеспечивающей защиту систем обратного осмоса за счет минимизации содержания катионов кальция и магния в воде; процесс умягчения осуществляется на фильтрах умягчения (фильтрах Na-катионирования) стадия корректировки значения рН с помощью комплекса пропорционального дозирования растворов кислоты или щелочи. Качество воды регламентирует нормативно-техническая документация: ГОСТы, Санитарные правила и нормы 2.1.4.1074-01, фармакопейные статьи на воду очищенную (ФС 42.2619.97). Согласно этим нормативным документам, в воде очищенной не допускается присутствие более 100 клеток микроорганизмов в 1мл воду. ЗаключениеВалидация в настоящее время является очень важным аспектом обеспечения качества ЛС. Сложное оборудование, многостадийный производственный цикл и высокие требования к качеству препаратов обуславливают жёсткие требования к стандартности (постоянству) условий производства в рамках GMP. Валидация каждого этапа производства KC, а также всего процесса в целом, позволяют обеспечить надлежащее качество выпускаемой на фармацевтическом предприятии продукции, а также постоянство таких характеристик лекарственного средства как биодоступность и биоэквивалентность на всём жизненном цикле (период производства) лекарственного препарата. Было установлено, что процесс валидации является необходимой составляющей на любой производстве. Процесс помогает выявить все проблемные точки и характеристики процесса и оборудования, показать несоответствие продукции или услуги заданным требованиям. Это может помочь улучшить все производство, наладить должным образом все процессы, увеличить качество выпускаемой продукции, что, в конечном счете, приведет к экономической выгоде. Валидация механических фильтров играет большую роль в процессе производства. Было доказано, что фильтры и их регулярная проверка является основополагающим элементом в процессе производства лекарственных средств. Воздушные механические фильтры устанавливаются для очистки воздуха в помещениях. Это препятствует проникновению с воздухом мелких и крупных загрязнений и микроорганизмов, которые, с проникновением в рабочий материал, могут изменить его структуру и характеристики, оказав влияние на качество выпускаемых лекарственных средств, а также на качестве жизни потребителя. Фильтры для очистки воды находятся на каждой стадии производства лекарственных средств. Очищение происходит на предварительном этапе, где очищенную воду использую для санитарных нужд, и на дальнейших этапах водоподготовки, где используются более тонкие фильтры. Вода, полученная на последних стадиях очистки, используется для производства ЛС. Существует довольно серьезная разница между российским и международным подходами к процедуре валидации. В российских правилах GMP, валидация «заключается в документированном подтверждении соответствия оборудования, условий производства, качества сырья и готового продукта действующим регламентам и (или) требованиям нормативной документации». Из разных публикаций может создаться впечатление, что процесс валидации является не более чем процессом документирования или, что такие слова как «валидация» «верификация», «квалификация» и «испытание», в самом деле, являются аналогами друг друга. В связи с этим, необходимо более серьезное и углубленное изучение данного вопроса. Важно особое внимание уделять процессу валидации как оборудования, так и продукции, услуг и методик, для более точного выявления проблемных вопросов производства и дальнейшего их устранения. Это поможет увеличить качественные услуги на рынке и будет способствовать экономическому росту в стране Используемая литератураАбрамова Е., Алексеева Н., Егоров А. Аттестация/квалификация (валидация) оборудования и аналитических методов в фармацевтическом производстве // Аналитика. 2012. – № 1. – с. 60 – 62. ГОСТ Р ЕН 1822-1-2010 Высокоэффективные фильтры очистки воздуха ЕРА, HEPA и ULPA. М.: Стандартинформ, 2011 Аладышева Ж.И. Основные принципы проведения валидации на фармацевтическом производстве/ Ж. И Аладышева, А. П. Мешковский, JI. М. Левин; под редакцией В.В. Береговых – М., 2005. – 186 с. Куликов Н.И., Найманов А.Я., Омельченко Н.П., Чернышев В.Н. Теоретические основы очистки воды. Донбасская национальная академия строительства и архитектуры, Макеевка, 2009. - 297 с Беляев В.В. Валидация в производстве и контроле качества лекарственных средств // Тезисы докладов межвузовской научной конференции студентов и молодых ученых "Фармация в XXI веке: Эстафета поколений". – С-Пб., 2009. – 146 с.. Кипор С.Г., Люлина Н.В. Валидация действующего фармацевтического производства // Ремедиум. - 2012. - № 7-8. - С. 78 - 80 Гармонов, С.Ю. Контроль качества и безопасность лекарственных препаратов / С.Ю. Гармонов. – Казань: КГТУ, 2008. – 324 с. ГОСТ Р 52249-2009. Правила производства и контроля качества лекарственных средств. Введ. 2009-06-08. – М.: Изд-во стандартов, 2009. – 132 с. Мешковский, А.П. О концепции внедрения правил НАДЛЕЖАЙЩАЯ ПРОИЗВОДСТВЕННАЯ ПРАКТИКА в России / А.П. Мешковский // Фарматека. 2003. – № 5. – с. 32–37. ИСО 9000:2001 Системы менеджмента качества. Основные положения и словарь. М.:ГосСтандарт России, 2001 Мешковский А.П. Правила НАДЛЕЖАЙЩАЯ ПРОИЗВОДСТВЕННАЯ ПРАКТИКА для вспомогательных веществ / А.П. Мешковский // Фарматека. – 1998. – №6. – с. 37–41. Попов А.Ю. Валидация критических процессов и зон / А.Ю. Попов // Чистые помещения и технологические среды. 2005. – № 2. – с. 11–13. Попов А.Ю. Как наиболее эффективно соответствовать требованиям GMP? / А.Ю. Попов // Чистые помещения и технологические среды. -2005. № 1. – с. 9–10. ГОСТ Р ИСО 9000-2011 Системы менеджмента качества. Основные положения и словарь. М.: Стандартинформ, 2012 Попов А.Ю. Система анализа риска как первый шаг в переходе к работе по правилам надлежащей производственной практики (GMP) / А.Ю. Попов, А.П. Мешковский // Фарматека. 2002. – № 4. – с. 62–64. Производство лекарственных средств. Валидация. Основные положения: методические указания / сост. ГУП "ГипроНИИмедпром", ФГУП ГНЦА; Минпромнаука России. М., 2003. – 10 с. Вальдберг А.Ю., Александров В.П. Фильтры для очистки промышленных газов. Учебное пособие. М.: МГУИЭ, 2009. - 204 с Руководство Правила надлежащего производства лекарственных средств для медицинского применения и для ветеринарного применения Таможенного союза (правила надлежащей производственной практики – Good Manufacturing Practice – GMP) Проект (по состоянию на 01 февраля 2013 г.) – М.: Ремедиум, 2012. – 264 с. ГОСТ Р ЕН 1822-1-2010 Высокоэффективные фильтры очистки воздуха ЕРА, HEPA и ULPA. М.: Стандартинформ, 2011 Шилова C.B. Валидация на фармацевтических предприятиях России / C.B. Шилова // Основные аспекты валидации: материалы научно-практического семинара / ГНЦА, Учебно-производственный центр GMP. – М., 2002. – с. 23–25. Шилова C.B. Организация проведения валидации на фармацевтическоп предприятии / C.B. Шилова // Основные аспекты валидации: материалы научно-практического семинара/ ГНЦА, Учебно-производственный центр GMP. М., 2002. – с. 25–27. Перекрестное загрязнение в химико-фармацевтическом производстве: проблемы стандартизации и унификации требований /Гармонов С.Ю, Нурисламова Г.Р, Фатхуллин Р.Р.//Актуальные проблемы современности. М.:, 2012 ФС 42-2619-97 Вода очищенная. М.: 1997 Квалификация и валидация. Что ждать от новых изменений GMP// Квартальные среды. 2014. №2. С.6-9 Руководство по качеству воды для применения в фармации [Электронный ресурс] // Росздравнадзор. URL: http://www.consultant.ru (дата обращения: 22.04.2015) Шилова С. В. Процесс очистки производственного оборудования и его валидация Электронный ресурс. / C.B. Шилова. режим доступа: http://www.gmp-club.com/ru/intersite/validation/val0106.html http://gmpnews.ru/2012/08/praktika-validacii-processov-na-primere-farmacevticheskoj-otrasli/ Документы для валидации СВП [Электронный ресурс] // ЗАО "НПК Медиана-Фильтр" [Офиц.сайт] URL: http://www.mediana-filter.ru (дата обращения: 25.05.2015) |