Ароматические углеводороды в нефтехимическом синтезе. Содержание Введение 4 1 Теоретическая часть 4 1 Химические свойства ароматических углеводородов 5 2 Использование ароматических углеводородов в нефтехимических
 Скачать 1.05 Mb. Скачать 1.05 Mb.
|

Приборы, реактивы, материалыПробирка с муфтой объемом 15 мл, стакан из термостойкого стекла вместимостью 750— 1000 мл, термометр с длинной ножкой, анилин свежеперегнанный марки ч., пипетки на 2 мл.
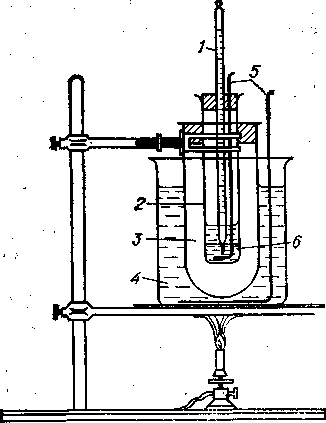
Метод равных объемов. В чистую и сухую пробирку 2 (рис. 7) помещают по 2 мл анилина и анализируемой бензиновой фракции, плотно закрывают пробкой со вставленным в нее термометром 1 и мешалкой 5 и укрепляют в муфте 3, погруженной в водяную баню 4. Продукт и анилин берут с помощью пипеток вместимостью 2 мл. Термометр помещают так, чтобы середина ртутного шарика находилась на уровне линии раздела слоев анилина и продукта. Температуру водяной бани медленно повышают, при этом непрерывно перемешивают мешалкой продукт с анилином. Отмечают температуру полного смешения жидкостей (при этом раствор становится прозрачным), прекращают нагревание и дают воде медленно остывать. Когда в пробирке 2 появляется муть, что свидетельствует о начале разделения фаз, снова начинают перемешивать раствор мешалкой. Вначале при перемешивании муть исчезает, но затем наступает момент неисчезающего помутнения. За анилиновую точку принимают наивысшую температуру, при которой муть при перемешивании не исчезает. Температуры полного смешения и помутнения не должны расходиться более чем на 0.10С. Определение анилиновой точки повторяют с новым образцом исследуемой фракции. Расхождение анилиновых точек в параллельных опытах не должно превышать 0.20С. Метод максимальной анилиновой точки (истинной КТР в анилине). В пробирку 2 помещают 2 мл исследуемой фракции, 1.6 мл анилина и определяют температуру полного растворения, как описано выше. После этого к смеси добавляют еще 0.2 мл анилина и снова определяют температуру растворения. Обычно она бывает выше, чем при первом определении. Анилин прибавляют по 0.2 мл до тех пор, пока после некоторого максимума температуры растворения не наметится ее снижение. Максимальную анилиновую точку фиксируют; она соответствует истинной критической температуре растворения исследуемого продукта в анилине. При наличии достаточного количества вещества для каждого определения следует брать новые порции продукта и анилина. Адсорбционный анализ основан на способности адсорбентов избирательно извлекать из смесей соединения определенных типов. Для разделения углеводородов применяют различные адсорбенты: окись алюминия, активированный уголь, силикагель и др. Чаще всего используют силикагель. Ароматические углеводороды более прочно удерживаются на поверхности адсорбента, чем парафиновые и нафтеновые. Пропуская смесь углеводородов сверху вниз по колонке с адсорбентом, обычно с добавлением растворителя, выделяют из колонки вначале парафиновые и нафтеновые углеводороды, а затем ароматические. Для выделенных фракций измеряют объем и исследуют (определяют наличие ароматических углеводородов, показатель преломления, анилиновую точку и т.п.). При адсорбционном разделении бензиновых фракций применяют два типа растворителей: вытесняющие и смещающие. Вытесняющие растворители — спирты (изопропиловый, этиловый, метиловый) — адсорбируются сильнее компонентов бензина и выделяются из колонки вслед за ароматическими углеводородами. В этом случае нельзя достигнуть полного разделения бензина на две фракции— парафино-нафтеновую и ароматическую, так как они движутся по колонке вплотную друг к другу. Поэтому приходится еще отбирать промежуточную фракцию, представляющую собой их смесь. Смещающие (разбавляющие) растворители — пентан, изопентан — близки по адсорбируемости к парафино-нафтеновой фракции. Такие растворители смешиваются в колонке с углеводородами, постепенно десорбируя их и заставляя двигаться вниз. Если вслед за смещающим растворителем (изопентан) ввести в колонку вытесняющий (метанол, этанол), то можно отделить парафино-нафтеновую фракцию без отбора промежуточной. Измеряя показатель преломленияфильтрата, можно обнаружить компоненты смеси в такой последовательности: парафины + нафтены —> парафины + нафтены + изопентан —> изопентан + ароматические углеводороды -> метанол + ароматические углеводороды —> метанол. Фракции парафино-нафтеновых и ароматических углеводородов выделяют из фильтрата отгонкой изопентана. Фракцию ароматических углеводородов отделяют от метанола промывкой водой, после чего обезвоживают СаСl2 и металлическим натрием. Для бензинов, содержащих до 15 объемных % ароматических углеводородов, удобно применять адсорбционное разделение с вытесняющим растворителем и отбором промежуточной фракции, при более высоком содержании в бензине ароматических углеводородов рекомендуется разделение при помощи смещающей жидкости + вытесняющий растворитель. Приборы, реактивы, материалыИсследуемый нефтепродукт или смесь, стеклянная колонка (рис. 3) высотой 200 мм, диаметром 8—10 мм с воронкой для подсадки пробы, рефрактометр типа ИРФ, изопропиловый спирт или пентан (изопентан), адсорбент – Al2O3,, мерные цилиндры (градуированные пробирки) с ценой деления 0.1 мл. Проведение анализа. Колонку заполняют сухим адсорбентом, укрепляют в штативе, наверху закрепляют делительную воронку 1 для подсадки пробы и элюента, под нижний конец колонки 2 подводят градуированную пробирку 3.
Исследуемую фракцию в количестве 5 мл заливают в колонку и после того, как она полностью впитается в адсорбент, добавляют в качестве десорбирующей жидкости 15-20 мл изопропилового спирта. Сначала из колонки будет выходить насыщенная (алкано-циклоалкановая) часть исследуемой фракции, которая адсорбируется Al2O3 менее прочно. Первую порцию отбирают в количестве 1 мл, а все последующие — по 0.5 мл. Для каждой отобранной фракции определяют показатель преломления. Фракции, отличающиеся по показателю преломления не более чем на 0.0005 смешиваются. Появление аренов замечают по формалитовой реакции: в пробирку помещают 1 мл 98 %-й бесцветной серной кислоты, добавляют 2—3 капли 10 %-го раствора формалина и столько же продукта. При отсутствии аренов смесь остается бесцветной или слегка желтеет. Ярко-красное окрашивание указывает на появление в отобранной фракции аренов. Удаление аренов серной кислотой Аппаратура, реактивы, материалы Сульфатор (рис. 4) Воронка с оттянутым концом, серная кислота. 98%-я Благодаря наличию градуированной трубки сульфатор может служить не только для удаления, но и для определения объемной доли аренов (и алкенов) в процентах. 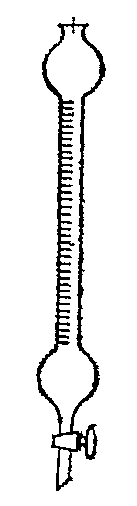 Рис. 4 – Сульфатор Рис. 4 – Сульфатор Хорошо вымытый и высушенный сульфатор закрепляют вертикально в штативе, закрывают кран и при помощи воронки с удлиненным концом осторожно наливают в него 98%-ю серную кислоту (так, чтобы она не растекалась по стенкам сульфатора) до отметки «О». Кислоте дают стечь со стенок и точно замечают ее уровень по шкале сульфатора. После этого таким же путем в сульфатор наливают 10 мл исследуемой фракции бензина и фиксируют его уровень. Сульфатор плотно закрывают пробкой, вынимают из штатива и осторожным наклоном переводят его содержимое в верхний шарик, где тщательно взбалтывают в течение 1 мин, время от времени приоткрывая кран для выпуска образовавшихся газов. По окончании перемешивания сульфатор устанавливают вертикально в штативе и дают смеси отстояться 1 ч. По истечении этого времени отмечают объем бензина. Определение следует повторить два раза и взять среднее арифметическое из результатов двух опытов. Объемную долю в исследуемой фракции аренов Ао, %, находят по формуле: А0 = [(V1 - V2)/V1) ∙ 100 где V1 — объем бензина, взятый на сульфирование, V2 — объем бензина после сульфирования. После измерения объема бензина, оставшегося после сульфирования, сернокислотный слои сливают, а бензин промывают в сульфаторе водой, затем раствором соды и снова водой, после чего переносят в сухую пробирку и обезвоживают прокаленным хлоридом кальция. Обезвоженный бензин берут для определения второй анилиновой точки. При необходимости получить большее количество деароматизированиой фракции обработку продукта серной кислотой можно проводить в делительной воронке, куда помещают исследуемую фракцию и двукратное (по объему) количество 98 %-й серной кислоты. Смесь в делительной воронке взбалтывают в течение 20 мин, время от времени приоткрывая кран для выпуска газа. При обработке легкокипящих или богатых аренами углеводородных фракций делительную воронку со смесью бензина и серной кислоты вначале охлаждают в бане со льдом в течение 15 мин и только после этого начинают взбалтывание. Если при взбалтывании смесь разогревается, ее следует вновь охладить. После окончания перемешивания смеси дают отстояться до полного расслаивания, кислотный слой спускают из воронки, а бензин промывают водой, 10%-м раствором гпдроксида натрия, а затем снова водой до нейтральной реакции. Тщательно отделив от воды, бензин переносят в сухую колбу и высушивают прокаленным хлоридом кальция. Определение индивидуального состава этой фракции проводят методом газожидкостной хроматографии на капиллярной колонке. Для анализа используют хроматограф с ионизационно-пламенным детектором. В качестве газа-носителя можно использовать азот, гелий, аргон, водород. Металлическая колонка (медная, стальная и т. п.) имеет длину 50 или 100 м и внутренний диаметр 0,25—0,3 мм. Затем колонку помещают в термостат и продувают газом-носителем при температуре, близкой к максимальной температуре хроматографического опыта. При определении состава ароматических углеводородов широкой фракции н. к. — 200 °С в качестве неподвижных жидких фаз используют полярные вещества, например полиэтиленгликоль (ПЭГ), дибутилтетрахлорфталат (ДБТХФ), трикрезилфосфат и др. В табл. 2.3 приведены значения относительного времени удерживания аренов на двух неподвижных жидких фазах. Идентификацию хроматографических пиков проводят с помощью индивидуальных аренов. Температура хроматографической колонки во время анализа 100 °С [5]. Заключение В курсовой работе были описаны химические свойства ароматических соединений, содержащихся в нефтях и нефтепродуктах, их распределение в различных фракциях нефти, влияние аренов на свойства нефтепродуктов. Рассмотрены методы удаления аренов из нефтяных фракций. Литература Поконова Ю.В., Гайле А.А., Спиркин В.Г. и др. Химия нефти / Под ред. З.И.Сюняева. Л.: Химия, 1984. 360 с. Рябов В.Д. Химия нефти и газа. М.: ГАНГ, 1998. 370 с. Богомолов А.И., Гайле А.А., Громова В.В. и др. Химия нефти и газа / Под ред. В.А. Проскурякова, А.Е. Драбкина. 2-е изд., перераб. Л.: Химия, 1989. 424 с. Абросимов А. А. "Экология переработки углеводородных систем". М: Химия, 2002. И.Н.Дияров и др. «Химия нефти» руководство к лабораторным занятиям,Ленинград «Химия» 1990г. |