Субхонова С.301А.сам.раб.фармхимия. Современные методы анализа антибиотиков
 Скачать 5.42 Mb. Скачать 5.42 Mb.
|

2.4 Количественное определение некоторых антибиотиков2.4.1 ПенициллинЙодометрический метод Под влиянием щелочей или пенициллиназы происходит раскрытие бета-лактамного кольца в структуре антибиотика с образованием пенициллоиновой кислоты. Последняя и реагирует с йодом. По разности йода, израсходованного на взаимодействие с пенициллином до и после гидролиза молекулы антибиотика, судят о количестве препарата в исследуемых образцах. Этот метод даёт хорошо совпадающие результаты с данными биологических методов определения активности, и он применим для большинства лекарственных форм пенициллина. Взвешивают 25-30 мг исследуемого образца антибиотика и растворяют в фосфатном буфере (рН 6,0) до получения концентрации примерно 2000 ЕД/мл. По 2 мл полученного раствора вносят в две 125-миллилитровые колбы Эрленмейера. В первую из них добавляют 2 мл 1N раствора едкого натра и оставляют на 15 мин при комнатной температуре. После этого в данную колбу вносят 2 мл 1,2 N раствора соляной кислоты и 10 мл 0,01 N йода. Через 15 мин избыток йода оттитровывают 0,01 N раствором гипосульфита натрия. В конце титрования добавляют 1 каплю раствора крахмала и продолжают титрование при энергичном встряхивании до обесцвечивания синей окраски крахмала. Во вторую колбу добавляют 10 мл 0,01 N раствора йода и титруют сразу 0,01 N раствором гипосульфита натрия (контроль). Активность пенициллина определяют по нижеприведённой формуле: 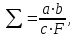 где Σ − количество ЕД пенициллина в 1 мг; a − разность в титрах; b − активность рабочего стандарта в ЕД/мг; c − вес образца, мг; F − количество мл 0,01N раствора йода, поглощённого 1 мл рабочего стандарта натриевой соли бензилпенициллина. Следует помнить, что расход йода при титровании зависит от температуры, рН среды, концентрации йода и других причин. Поэтому необходимо строго соблюдать условия стандартности опытов, тогда результаты будут неизменно воспроизводимыми. Весовой метод определения бензилпенициллина Этот метод является ориентировочным, так как характер и количество осадка зависят от условий осаждения. Бензилпенициллин с N-этилпиперидином образует кристаллические соли, плохо растворимые в смеси ацетил-ацетона и при определённой концентрации выпадающие из раствора в виде осадка. Это позволяет определить пенициллин G не только в виде кристаллической натриевой соли, но и в смеси с другими солями пенициллина. Метод не пригоден для определения пенициллина G в смеси с пенициллином X. Метод ультрафиолетовой спектрофотометрии Фенильная группа в пенициллине G позволяет получать характерный максимум поглощения в ультрафиолетовом свете при 263 нм. Хроматографичевкий метод Смесь пенициллинов достаточно чётко разделяется на полосках хроматографической бумаги. Проявление местоположения их осуществляется по зонам задержки роста тест-микроба вокруг определённых участков полоски, наложенной на микробный газон. Метод выполняется следующим образом. Полоски хроматографической бумаги (35×0,5 см) пропитываются раствором фосфатного буфера (рН 6,8), представляющего смесь насыщенных растворов дигидрофосфатов калия и натрия, и подсушивают между слоями фильтровальной бумаги при окончательном досушивании на воздухе. На линию старта наносят по 1-2 мл испытуемого раствора с исходной концентрацией 800-1000 ЕД/мл. Бумагу помещают в хроматографическую банку, в лоток которой налит освобождённый от перекисей эфир, насыщенный водой (нисходящая хроматография). Банку ставят в рефрижератор на сутки. После этого бумажную ленту отрезают на 0,5 см выше линии старта и переносят на засеянные тест-микробами лотки. Лента должна плотно прилегать к агару. Лотки оставляют в термостате (37º) на 16-18 ч и затем читают результаты. Количество зон задержки роста культуры соответствует числу типов пенициллинов, а порядок чередования их на бумажной ленте зависит от коэффициента распределения. Наиболее подвижен пенициллин К (гептилпенициллин), за ним последовательно располагаются дигидропенициллин F (амилпенициллин), пенициллин F (2-пентенилпенициллин), пенициллин G (бензилпенициллин) и пенициллин Х (пара-оксибензилпенициллин0. Чем больше зона задержки роста, тем больше в смеси того или иного типа антибиотика, количество которого можно определить следующим образом: отрезанную бумажную ленту разрезают по длине на две равные части, одну из которых помещают на агар с тест-культурой, а вторую через 16-18 ч разрезают на части соответственно зонам задержки роста. Пенициллин из обрезков извлекают водой и водные экстракты титруют чашечным методом. Этот способ менее точен, чем описанный ранее диффузионный, а поэтому его следует расценивать лишь как ориентировочный. Тем не менее качественное определение пенициллинов хроматографическим методом распространено широко. 2.4.2 СтрептомицинМальтольный метод Этим методом возможно определение не только стрептомицина, но и маннозидстрептомицина. Он основан на том, что оба названных антибиотика при нагревании с раствором разбавленной щёлочи образуют мальтол, который определяется затем колориметрически или спектрофотометрически по окраске, образуемой им с солями железа или реактивом фолина. В мерную колбу на 25 мл наливают 10 мл испытуемого образца стрептомицина-основания, содержащего 0,5 мл антибиотика и 1 мг раствора. В эту же колбу добавляют 2 мл 1 N NaOH и ставят её на кипящую водяную баню на 10 мин. Затем охлаждают колбу в ледяной или проточной водопроводной воде и приливают 2 мл 1,2 N HCl; добавляют 5 мл 0,25% хлористого железа и доводят общий объём смеси дистиллированной водой до метки. Полученный окрашенный раствор калориметрируют при 550 нм в соответствующем фотоэлектрокалориметре против раствора стандартного образца антибиотика и дистиллированной воды в контролях. Результаты выражают графически на полулогарифмической сетке, где по оси ординат откладывают значения для степени прохождения света, а по оси абсцисс − концентрацию стрептомицина. Активность испытуемого образца вычисляют по стандартной кривой. Недостатки метода: нельзя определять стрептомицин и маннозидстрептомицин отдельно при нахождении их в смеси; если в последней будет ещё и оксистрептомицин, то он в условиях опыта даст оксимальтол, который также образует окрашенные продукты с солями железа. Нитропруссидный метод Этим методом можно определять различные стрептомицины, так как известно, что нитропруссид натрия в щелочной среде даёт окрашивание с гуанидином. Определение выполняется следующим образов: смешивают равные объёмы 10% растворов нитропруссида натрия, железистосинеродистого калия и едкого натра. К смеси приливают 3 объёма воды. Через 30 мин раствор готов к употреблению (он имеет ярко-жёлтую окраску). При появлении мути в растворе его заменяют свежим. Обычно реактив пригоден для работы лишь в течение нескольких часов. К 5 мл раствора стрептомицина (концентрация 20-25 Ед/мл) приливают 1 мл реактива. При этом возникает устойчивая оранжевая окраска. Раствор калриметрируют против раствора стандартного образца стрептомицина (контроль). Последний должен быть прозрачным, поскольку мутные растворы могут исказить результаты определения. Метод не вполне специфичен, так как окрашенные соединения получаются и с другими веществами, содержащими гуанидиновые группы, и, в частности со стрептидином − продуктом расщепления стрептомицина, не обладающим биологической активностью. Микроманометрический метод Метод применим для определения низких концентраций антибиотика (0,005-0,05 ЕД/мл). В сосудики от аппарата Варбурга вносится взвесь 16-18-часовой культуры золотистого стафилококка 209Р (25-100 млн. клеток/мл), мясо-пентонный бульон без NaCl и глюкозы; рН среды 8,0. Объём смеси в каждом сосудике должен быть равен 2 мл. В центральную часть сосудика наливают 0,2 мл 1 N раствора NaOH. Температура водяной бани − 37º. Стрептомицин используется в концентрациях от 0,005 до 0,05 ЕД/мл. При этом между количеством поглощённого бактериями кислорода и концентрацией стрептомицина существует прямолинейная зависимость. Измерение дыхания проводят в пределах 2,5 ч с интервалом измерений объёма поглощённого кислорода через каждые 15-30 мин. По результатам опытов строят стандартную кривую, на основании которой производят расчёт активности в испытуемом образце. Для этого по оси ординат откладываколичество (мм3) поглощённого кислорода за 2,5 ч, а по оси абсцисс − концентрацию антибиотика (ЕД/мл). Следует отметить, что растворы стрептомицина более высокой концентрации, чем 0,05 ЕД/мл, необходимо предварительно разбавлять до концентрации 0,005-0,05 ЕД/мл или определять другими методами. Микроманометрическое определение пригодно для выявления концентрации стрептомицина в негемолизированной крови (гемолизированная кровь убивает стафилококка). 2.4.3 ТетрациклиныКолориметрический метод Фенольно-гидроксильные группы в структуре хлортетрациклина, окситетрациклина и тетрациклина позволяют использовать цветную реакцию их с хлорным железом для количественного определения названных антибиотиков. В колбу Эйленмейера (объёмом 50 мл) вносят 10 мл раствора испытуемого препарата − основания, приготовленного путём растворения последнего в 0,01 N HCl и содержащего 0,1 мг антибиотика в 1 мл. В эту же колбу наливают 10 мл 0,05% раствора хлорного железа, перемешивают и оставляют при 16-18º на 10 мин, после чего определяют прохождение света для стандарта и испытуемого препарата при 490 нм на ФЭК против 0,01 n раствора соляной кислоты (контроль). По данным анализа для растворов стандарта строят стандартную кривую на полулогарифмической сетке (по оси ординат откладывают показатели колориметра, а по оси абсцисс − концентрацию антибиотика-основания). По этой кривой определяют количество антибиотика в испытуемом образце. Подобным методом возможно определение хлортетрациклина и тетрациклина, приобретающих стойкую жёлтую окраску при нагревании с соляной кислотой. Окситетрациклин такой реакции не даёт. Спектрофотометрический метод Растворы окситетрациклина и тетрациклина дают жёлтое окрашивание при добавлении щёлочи. При этом максимальное поглощение выявляется при 380 нм. Хлортетрациклин при обработке щёлочью даёт нестойкое жёлтое окрашивание и, по существу, не удаётся выявить максимум при 380 нм. Следовательно, данный метод позволяет определять окситетрациклин и тетрациклин в присутствии хлортетрациклина. Описаны и другие методы определения тетрациклинов и, в частности, хроматографические, флуорометрический и пр. 2.4.4 ЛевомицетинМетод диазотирования с последующим титрованием 0,1 М раствором нитрита натрия (ГФХ). Берут точную навеску препарата (0,5 г) и помещают её в коническую колбу на 250 мл, приливают 20 мл концентрированной соляной кислоты и осторожно, небольшими порциями, 5 г цинковой пыли. Затем приливают ещё 10 мл той же кислоты, обмывая стенки колбы, и после полного растворения цинковой пыли (можно подогреть), раствор количественно переносят в стакан для диазотирования, охлаждаемой снаружи льдом; прибавляют 3 г бромида калия и медленно титруют 0,1 M раствором нитрита натрия. Титрование считают законченным, когда капля жидкости, взятая через 3 мин после прибавления раствора нитрита натрия, будет вызывать немедленное посинение йодкрахмальной бумаги; 1 мл 0,1 М раствора NaNO2 соответствует 0,03231 г левомицетина. Колориметрический метод Этот метод основан на восстановлении нитрогруппы левомицетина в аминогруппу, которая при диазотировании образует окрашенные соединения. В колбу Эрленмейера на 10 мл вносят 2 мл раствора испытуемого образца в дистиллированной воде, содержащего 15 мкг антибиотика в 1 мл. Затем добавляют 1 мл 0,3 N NaOH и 25 мг NaHSO3. Полученный раствор оставляют при 16-18º в течение 15 мин. Спустя указанный срок в колбу приливают 0,5 мл 5 % раствора NaNO2 и 5-10 капель концентрированной (36-37%) соляной кислоты. Через 1-3 мин добавляют 1 мл 5% раствора сульфаминовой кислоты. Образующиеся в колбе азотистые пары удаляют с помощью водоструйного насоса в течение нескольких секунд. Затем приливают0,5 мл 0,5% раствора N-(1-нафтил)-дихлордиэтилендиамина и доводят объём дистиллированной водой до метки. Через 2 ч производят замеры на ФЭК при 558 нм против дистиллированной воды, обработанной вышеуказанным способом (контроль). Концентрацию испытуемого образца определяют по стандартной кривой на полулогарифмической сетке. Указанный метод чаще используется при определении хлорамникола, получаемого ферментативным путём. 2.4.5 ЭритромицинСпектрофотометрический метод Этот метод позволяет определять эритромицин даже в присутствии продуктов его кислой инактивации (в отличие от колориметрических методов). Он основан на измерении оптической плотности раствора эритромицина при 236 нм после слабой щелочной обработки. Для внесения поправки на поглащение света различными примесями и продуктами разложения используют раствор эритромицина, инактивированный кислотой (контроль). 2.4.6 НеомицинКолориметрический метод Метод основан на количественном определении пентозы, входящей в структуру антибиотика. К 1 мл испытуемого раствора антибиотика добавляют 2 мл концентрированной соляной кислоты с хлорным железом (100 мг FeCl3 на 100 мл HCl) и 0,1 мл 10% раствора орцина в этаноле; затем смесь нагревают на водяной бане в течение 20 мин и быстро охлаждают; объём доводят дистиллированной водой до 10 мл и фотометрируют на ФЭК с красным светофильтром (610-750 нм). Расчёт количества неомицина в испытуемом образце ведётся по стандартной кривой, построенной по данным измерения на ФЭК растворов стандарта антибиотика. 2.4.7 ФлоримицинКолориметрический метод Метод основан на определении антибиотика по биуретовой реакции. Точную навеску флоримицина (80-90 мг) растворяют в 100 мл дистиллированной воды в мерной колбе. Полученный раствор в количестве 20 мл переносят в 50-миллилитровую колбу Эрленмейера, добавляют 2 мл 2% раствора CuSO4×5H2O и 5 мл 2 N раствора NaOH. Содержимое колбы доводят до метки дистиллированной водой и тщательно перемешивают. Спустя 3 мин выпавший осадок гидроокиси меди отфильтровывабт через бумажный фильтр. После этого измеряют оптическую плотность прозрачного фильтрата, окрашенного в фиолетовый цвет. Используют ФЭК с зелёным светофильтром; длина кюветы 5 см. В качестве контроля используют дистиллированную воду. Расчёт активности испытуемого образца флоримицина ведётся на калибровочной кривой, построенной для стандартного флоримицина с известными концентрациями. Описанный метод применим для определения активностиантибиотика в культуральной жидкости. При этом, кроме указанных выше реактивов, используют смолу кБ-1 с содержанием дивинилбензола ≈6% или КБ-2 с содержанием дивинилбензола 2,5-3%; 2 N раствор NaOH и 0,5 N раствор H2SO4. 2.4.8 Грамицидин СМетод скоростной хроматографии на бумаге Грамицидин С избирательно проявляется бром-фенолом синим на бумажной хроматограмме. Это свойство было использовано для определения антибиотика на хроматограммах и в спиртовых экстрактах. Хроматографирование проводится в камере, сваренной из нержавеющей стали. Подставка изготавливается из стеклянных палочек. В качестве растворителя применяют метилэтилкетон – пропионовая кислота – вода (15:5:6) или н-бутанол-уксусная кислота – вода (4:1:5). Вторая система перед употреблением уравновешивается при 60º, для чего она выдерживается без отделения водного слоя при этой температуре не менее 30 мин. Используют верхний слой. На дно камеры помещают чашку Петри с системой растворителей. На бумагу наносят 10 или 25 мкл испытуемого раствора, содержащего в 1 мл 1000 мкг антибиотика (параллельно со стандартным образцом грамицидина). При содержании в образце более 5000 мкг/мл его необходимо предварительно развести, но не более чем до 50 мкг в 1 мл. Бумагу с нанесёнными образцами и стандартом заправляют в сухую хроматографическую лодочку, камеру плотно закрывают крышкой с зажимами и помещают её при 60º на 20-25 мин. По истечении указанного времени растворитель наливают через отверстие в крышке камеры (ранее закрытое резиновой пробкой) в лодочку. Отверстие закрывают и камеру оставляют при 60º на 30-60 мин. Затем хроматограмму вынимают, проявляют бромфеноловым синим и высушивают. Оценку результатов проводят с помощью денситометра. Возможно также элюирование окрашенных пятен спиртом при нагревании и их спектрофотометрии при 580 нм. 2.4.9 ЦиклосеринКолориметрический метод Метод основан на реакции циклосерина с нитропруссидным реагентом (смесь равных объёмов 4% раствора нитропруссида натрия и 4 N раствора едкого натра) в кислой среде с образованием комплекса голубого цвета. К 1 мл раствора циклосерина с концентрацией 50-200 мкг/мл добавляют 3 мл 1 N раствора уксусной кислоты и 1 мл приготовленного накануне нитропруссидного реагента. После перемешивания раствор оставляют при комнатной температуре нВ течение 10 мин и колориметрируют с красным светофильтром против дистиллированной воды (контроль). Длина кюветы 0,5 см. Концентрацию антибиотика рассчитывают по стандартной кривой, построенной для растворов стандарта. Колориметрическим методом можно определять циклосерин в присутствии продуктов его инактивации в кислой, щелочной и нейтральной средах. На результаты не оказывают влияние Na2SO4, MgSO4, (NH4)2SO4, NaCl и CaSO4 в концентрациях не выше 1,6 N. ЗаключениеГотовый антибиотик, полученный в форме вещества, подвергается химической и биологической проверке по всем пунктам, указанным в Фармакопее. Каждую партию антибиотика проверяют особо. Если препарат отвечает всем требованиям Фапмакопеи, то его затем превращают в лекарственные формы (т. е. расфасовывают в ампулы, если препарат предназначен для инъекций, таблетируют или помещают в капсулы, если препарат предназначен для приёма внутрь, и т. д.). лекарственные формы затем снова проверяют соответствующим методом, чтобы удостовериться, что препарат не испортился при превращении его в лекарственную форму и содержание антибиотика в каждой отдельной дозе (например, в одном флаконе или таблетке) отвечает указанной активности. Готовые антибиотики контролируют химически, микробиологически и фармакологически и проверяют клинически. Химический контроль предусматривает прежде всего определение содержания антибиотика с помощью некоторых химических методов, описываемых подробно в Фармакопее. Биологическая активность играет важнейшую роль в оценке химиотерапевтических свойств того или иного антибиотика, поэтому естественно стремление научных и практических работников применять микробиологический контроль в производстве, клинике и лаборатории. Однако в ряде случаев незаменимыми являются физико-химические методы определения активности антибиотических. В данной работе были рассмотрены методы, которые наиболее часто используются для анализа антибиотиков. Список использованной литературыГерольд М., Вондрачек М., Несачек Я., Доскочил И. Антибиотики. − Москва: "Медицина", 1966. Кашкин П. Н., Безбородов А. М., Елинов Н. П., Цыганов В. А. Антибиотики. − "Медицина", Ленинградское отделение, 1970. Государственная фармакопея СССР.- 11-е изд.- Вып.I, - М.: Медицина, 1987г. Блинов, Хохлов. Бумажная хроматография антибиотиков. Блок Р. Аналитические методы белковой химии. М 1963; 470. Дэвени Е., Гергей Я. Аминокислоты, пептиды и белки. М: Мир 1976. |