вв. Зантя_3_для_ст_методичка_измененная. Спадкові хвороби обміну. Факоматози. Онкогенетичні синдроми. Принципи лікування спадкових хвороб
 Скачать 276.5 Kb. Скачать 276.5 Kb.
|

1 2 V. Зміст теми заняття викладено у вигляді графічних схем. Схема 1. Граф логічної структури теми «Спадкові хвороби обміну».  Мутантний аллель Мутантний аллельЕтіологія Патогенез СХО  Патологічний первинний продукт Патологічний первинний продукт  Порушення ланцюжка біохімічних реакцій Порушення ланцюжка біохімічних реакцій    Патологія всередині клітини Патологія всередині клітиниПатологія органів Патологія організму Клінічні прояви     Порушення проміжного обміну Порушення проміжного обмінуПорушення синтезу та розпаду складних молекул Дефекти медіаторів та пов’язані з ними порушення  ТШХ, ВРХ амінокислот, вуглеводів ТШХ, ВРХ амінокислот, вуглеводів   ГХ-МС органічних кислот ГХ-МС органічних кислот    Тандемна масс-спектрометрія Тандемна масс-спектрометрія Скринінг-тест Ензимодіагностика Молекулярно-генетичне дослідження  На рівні субстрата На рівні субстрата     На рівні продукта гена На рівні продукта генаКорекція на рівні ферментів Хірургічне лікування Етіологічне лікування Схема 2. Граф логічної структури теми «Факоматози». 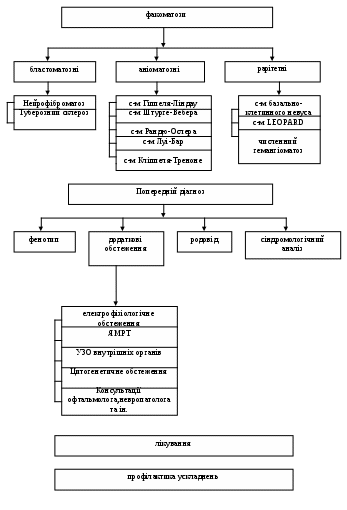 Схема 3. Граф логічної структури теми «Онкогенетичні синдроми». 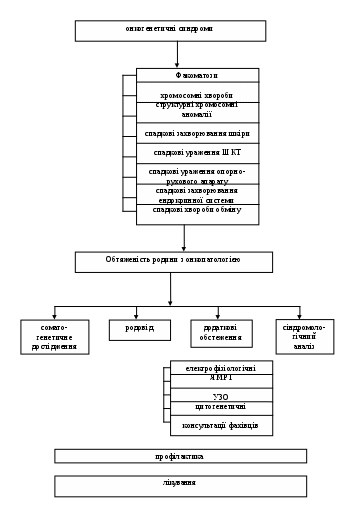 VI. План і організаційна структура заняття:
VІІ. Матеріали методичного забезпечення заняття: 7.1. Матеріали контролю для підготовчого етапу заняття. Теоретичні основи засвоєння теми «Спадкові порушення обміну речовин». Введення. Етіологія та патогенез спадкових хвороб обміну. Частота спадкових хвороб обміну. Загальні клінічні та біохімічні прояви. Класифікація спадкових порушень обміну речовин. Механізми запуску метаболічного кризу. Ролі сімейного анамнезу в діагностики метаболічних захворювань. Особливості семіотики спадкових порушень обміну речовин. Діагностика спадкових хвороб обміну. Етіологія, патогенез, клініка фенілкетонурії. Етіологія, патогенез, клінічна характеристика галактоземії, фруктоземії, глікогенозів. Етіологія, патогенез, клінічна характеристика мукополісахарідозів. Лізосомні хвороби накопичення; загальна характеристика. Принципи лікування спадкових порушень обміну речовин: симптоматичне, патогенетичне, хірургічне, етіотропне. Теоретичні основи засвоєння теми «Факоматози» та « Онкогенетичнісиндроми»: Введення. Загальна характеристика факоматозів, їх частота серед хворих зі спадковими ураженнями ЦНС. Онтогенетичних характер факоматозів. Етіологія, патогенез і класифікація факоматозів. Клінічний поліморфізм та генетична гетерогенність факоматозів. Бластоматозні форми факоматозів. Нейрофіброматоз. Класифікація. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Туберозний склероз. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Фруст-форми. Пренатальна діагностика. Ангіоматозні форми факоматозів. Синдром Луї-Бар. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Синдром Кліппеля-Треноне. Форми синдрому. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Пренатальна діагностика. Хвороба Штурге-Вебера. Форми хвороби. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Хвороба Гіппеля-Ліндау. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Хвороба рандю Ослера. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Раритетні форми факоматозів. Синдром нетримання пігменту (Блоха-Сульцбергера, гіпомеланоз ІТО). Класифікація. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Синдром Горліна-Горльца. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Синдром множинних лентіго. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Синдром Лешке. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Невус сальних залоз лінійний. Клініка, генетика, діагностика, тактика ведення хворих і членів їх сімей, прогноз. Частота, характер і термін маніфестації неопластичних уражень при факоматозах. Тактика ведення хворих на факоматози та членів їх сімей. Профілактика розвитку пухлин. Матеріали для тестового контролю (ІІ α): І. Тести з однією правильною відповіддю. 1.Які захворювання підлягають масовому біохімічному скринінгу: нейрофіброматоз; гемохроматоз; мукополісахаридози; фенілкетонурія 2. Які симптоми є показанням для проведення спеціалізованих біохімічних тестів: розумова відсталість, вроджені вади розвитку різних органів та систем; звичайне не виношування вагітності; катаракта, гепатоспленомегалія, відставання у розвитку; розторможення, порушення поведінки, імбецильність, незвичайний запах сечі. 3. Для якого ОГС характерна рабдоміома серця? хвороба Штурге – Вебера нейрофіброматоз глікогеном туберозний склероз синдром Вільямса 4. У хворого 2-х рокiв з "ляльковим" обличчям, низьким зростом, великим животом, збiльшенням розмiрiв печiнки вiдмiчаються натщесерце судоми. Приблизний дiагноз: Галактоземiя Мукополiсахаридоз Глiкогеноз Фукозидоз 5. Якi змiни спостерiгаються в клiтинах кiсткового мозку при сфінголіпідозах? a) збiльшення кiлькостi лiзосом b) зниження кiлькостi мiтохондрiй c) дефекти мембран лiзосом d) наявнiсть "пiнистих" клiтин 6. Зазначте тип успадкування переважної бiльшостi мукополiсахаридозiв? аутосомно-домiнантний аутосомно-рецесивний х-зчеплений рецесивно х-зчеплений домiнантний 7. У дитини з народження визначається вiдставання в ростi, деформацiї кiстяка, гiпертрихоз, розумова вiдсталiсть, гепатомегалiя. Попереднiй дiагноз: a) синдром Корнелiї де Ланге b) мукополiсахаридоз c) синдром Коффiна-Сiриса d) синдром Рубiнштейна-Тейбi e) глiкогеноз 8. З перших тижнiв життя клiнiчно манiфестує: фенiлкетонурiя галактоземiя мукополiсахаридоз пуриноз хвороба Вiльсона-Коновалова 9.Якщо в дитини спостерігається втрата набутих навичок або регресія нервово-психічного розвитку, вона має бути обстежена на: a) аміноацидурію b) хвороби накопичення c) хромосомні транслокації d) моногенни хвороби e) тератогенний вплив під час внутрішньоутробного розвитку 10. При яких захворюваннях спленомегалія є обов'язковим симптомом? a) мукополісахаридозах b) хворобі Гоше c) глікогенозах d) галактоземії 11. У дівчинки, 2 міс, що народилася вчасно від першої нормальної вагітності, батьки помітили різкий мишачий запах сечі. З порушенням метаболізму яких речовин це пов'язано? a) вуглеводів b) ліпідів c) амінокислот d) амонієвих сполук 12. Яке з нижченаведених тверджень є справедливим для фенілкетонурії? a) це аутосомно-домінантне захворювання b) однією з поширених ознак фенілкетонурії є гіперпігментація c) раннє призначення дієти (на 1-му місяці життя) дозволяє уникнути затримки розумового розвитку d)у дорослих обмеження в дієті можуть бути зняті e) у хворих з феніл кетонурією часто виявляються вроджені вади серця 13. Який переважний тип успадкування при спадкових захворюваннях обміну речовин? a) аутосомно-домінантний b) аутосомно-рецесивний c) зчеплений зі статтю d) мітохондріальний Матеріали методичного забезпечення для основного етапу заняття.
Завдання, які необхідно вирішити під час практичної частини заняття: Зібрати скарги, загальний анамнез та генетичний анамнез у пробанда. Розпізнати клінічні прояви спадкових хвороб. Призначити допоміжні лабораторні та інструментальні методи дослідження. Скласти родовід сім’ї, встановити механізм передачі спадкового захворювання. Ознайомлення з даними консультацій відповідних спеціалістів. Обґрунтувати діагноз. Скласти план лікування, профілактики, визначити прогноз хвороби. Провести об’єктивне обстеження хворого. Описати фенотип. Оцінити результати лабораторних та інструментальних методів обстеження, генетичних методів обстеження. Поставити попередній діагноз. Скласти план реабілітації та соціальної адаптації хворих. Матеріали контролю заключного етапу заняття. Нетипові задачі ІІІ рівня Задача 1. Дитина 6 років спрямована на консультацію з приводу частого головного болю. Фенотип: численні пігментні плями, пігментація в пахвовій області. У матері виявлені численні пігментні плями, нейрофіброми, пігментація в пахових областях. Батько таких змін не має. Ваш діагноз? Який ризик народження у батьків хворої дитини? Задача 2. Батьки дівчинки 6 років, звернулися за консультацією лікаря-генетика для уточнення діагнозу в зв'язку з затримкою психо-моторного розвитку. Особливості фенотипу: наявність ангіом на лівому стегні, ангіектазій на правій половині грудної клітки, правій руці. Права рука на 1 см довше лівої, ліва нога на 1,5 см довше правої. Також у дитини має місце правосторонній сколіоз. Ваш діагноз? Яка форма захворювання має місце? Задача 3. Дитина 4 років направлення на консультацію до лікаря-генетика у зв'язку з частими запальними захворюваннями верхніх дихальних шляхів. При огляді виявлено наявність телеангектазій на обличчі, ін'єкції судин кон'юнктиви, атаксія, гіпомімії особи. При додатковому обстеженні виявлено підвищений рівень хромосомної нестабільності (7%). Ваш діагноз? Який ризик при цьому захворюванні для сибсов? Задача 4. У молодих і здорових батьків народилася дитина, у якої на 6 день життя з'явилися блювота, понос. Вона відмовляється від годівлі. Іноді відзначається пітливість. Через якийсь час розвилася катаракта, збільшилася в розмірах печінка. Необхідно виключити спадкове захворювання: Задача 5. Дитина 3 роки госпіталізована в клініку з приводу епісиндрома, затримки психо-моторного розвитку. При огляді виявлено велику кількість депігментованих плям, в області щік - аденоматоз сальних залоз. У неврологічному статусі має місце двостороння пірамідна недостатність, команди не виконує, фразова мова відсутня. На ЕЕГ - підвищена судомна готовність. 1. Яке захворювання повинен запідозрити лікар? А. Нейрофіброматоз Б. Туберозний склероз. В. Синдром Луї-Бар. Г. Фенілкетонурія. 2.Якими додатковими дослідженнями можна підтвердити цієї діагноз? А. Огляд очного дна. Б. краніографія. В. Дослідження хромосомної нестабільності. Г. Ехографія внутрішніх органів. 3.Які основні напрямки лікування? А. Протівоссудорожная терапія. Б. Вітамінотерапія. В. Кардіотонічні і судинні засоби. Г. Хірургічне видалення пухлин. Задача 6. Дитина 3,5 років спрямована на консультацію до лікаря-генетика у зв'язку з частими епілептичними нападами. Мати дитини відзначила відразу після народження наявність у дівчинки ангіоми на лівій половині обличчя. З анамнезу відомо, що у віці 6 місяців у дитини розвинувся судомний пароксизм у вигляді джексонівського епіпріступу тривалістю 2 хвилини в правих кінцівках. Після чого відзначено розвиток генералізованих судомних нападів, які були резистентні до протисудомних препаратів. При додатковому обстеженні виявлено електроенцефалографічні ознаки підвищеної судомної готовності переважно в лівій лобно-скроневої області; на очному дні - розширення судин сітківки. 1. Ваш діагноз? А. Синдром Кліппеля-Треноне. Б. Фенілкетонурія. В. Синдром рандом Ослера. Г. Хвороба Штурге-Вебера. 2. Яка форма захворювання має місце? А. Класична тріада. Б. Нейрокутанеальная. В. моносімптомная. Г. безсимптомно. 3. Які принципи профілактики ускладнень цього захворювання? А. Профілактика інфекційних ускладнень. Б. Тривала дієтотерапія. В. Медико-генетичне консультування. Г. Дегідратаційних терапія. Задача 7. П'ятимісячна дитина поступила в клініку з приводу гіпотрофії, яка виникла на фоні постійних зригувань і блювання, що з'явилися відразу після народження. Під час об'єктивного обстеження виявлено гепатомегалію. Лабораторне дослідження встановило наявність анемії, гіпоглікемії, підвищення рівня прямого білірубіну. Ультразвукове дослідження органів черевної порожнини виявило збільшення розмірів нирок і печінки. Яке захворювання є причиною гепатонефромегалії? Задача 8. У клініку поступила дитина у віці 3 років. Дані огляду: низький зріст, кіфотична постава, короткі кінцівки, велика голова, грубі риси обличчя, сідлоподібний ніс, помутніння рогівки. Під час аускультації вислуховується систолічний шум уздовж лівого краю груднини й над верхівкою. Відзначається збільшений живіт, гепатоспленомегалія. Який діагноз найбільш імовірний? Матеріали методичного забезпечення самопідготовки студентів: орієнтовна карта для організації самостійної роботи студентів з навчальною літературою.
Література. Основна: Бочков Н.П. "Клінічна генетика". / Г. Медицина, 1997. Козлова С.І. "Преконцепційна профілактика вродженої патології"// Медико-генетичне консультування в профілактиці спадкових хвороб: Сб.матеріалів.-М., 1997. - 59-63 Після засвоєння необхідних знань вивчіть наступний матеріал: «Медична генетика» Під редакцією чл-кор. АМНУ, проф. Е. Я. Гречаніної, проф. Р.В. Богатирьової, проф. О.П. Волосовця. Підручник для студентів вищих медичних закладів ІІІ-ІV рівнів акредитації. Київ "Медицина" 2007. 534 с. Рекомендовано МОЗ України. Богатирьова Р.В., Гречаніна Е. Я. «Генетика репродуктивних втрат» - К., 2003. - 206 с. Молодан Л.В. Онкогенетичні синдроми// Ультразвукова перинатальна діагностика. - 2005. № 18. - С.113-148. 3. Гречаніна О.Я. "Проблеми сучасної генетики» Харків, 2003, 4 - 44 с. Гречаніна Е. Я., Колесников Д. Б. "Актуальні питання діагностики в клінічній генетиці" / Х. Т. Д.: 1995. - С. 129-148 Гречаніна Е. Я., Молодан Л. В., Бусигіна В. Ю. "Мезенхімальні дісплазії як одна з форм онкогенетічніх синдромів" Наследственные нарушения нервно-психического развития детей: Руководство для врачей/ Под ред. П.А.Темина, Л.З.Казанцевой.- М.:Медицина, 2001. – 432 с.:ил. Барашнев Ю.И. Перинатальная неврология. – М.: Триада-Х, 2001. – 640с. Додаткова: Бадалян Л.О. Детская неврология. - М., 1984. - 576 с. Барашнев Ю.И.,Вельтищев Ю.Е. Наследственные болезни обмена веществ у детей.-Медицина.,Ленинград, 1978. Ю.И.Барашнев, В.А.Бахарев, П.В.Новиков. Диагностика и лечение врожденных и наследственных заболеваний у детей». – М., «Триада-Х», 2004 г. The metabolic basis of inherited diseases/ Ed.D.B.Stanbury.-New York: McGraw-Hill. 1989. Мещишин І.Ф. Особливості обміну речовин у дітей. – Чернівці, 2003. – 108 с. 1 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||