Хроматография лаб. работа. Хроматография доклад. Теоретическое описание Определение хроматографии
 Скачать 2.08 Mb. Скачать 2.08 Mb.
|

|
Теоретическое описание Определение хроматографии по ИЮПАК – физический метод разделения веществ, в котором разделяемые компоненты разделяются между двумя фазами, одна из которых неподвижна, а другая движется в определенном направлении относительно первой. Все методы относятся к газовой хроматографии (ГХ) либо к жидкостной хроматографии (ЖХ). В ГХ разделяют вещества между неподвижной твердой и подвижной газообразной фазами. ЖХ разделяется на эксклюзионную (ситовую) хроматографию (основан на различиях в размерах частиц) и высокоэффективную ЖХ (основан на различиях в адсорбции или распределении веществ). В нашем опыте используется газовый хроматограф Кристалл 5000. В основе всех методов лежит процесс установления равновесия между неподвижной и подвижной фазами. Схематический рисунок процесса разделения компонентов в хроматографической колонке: 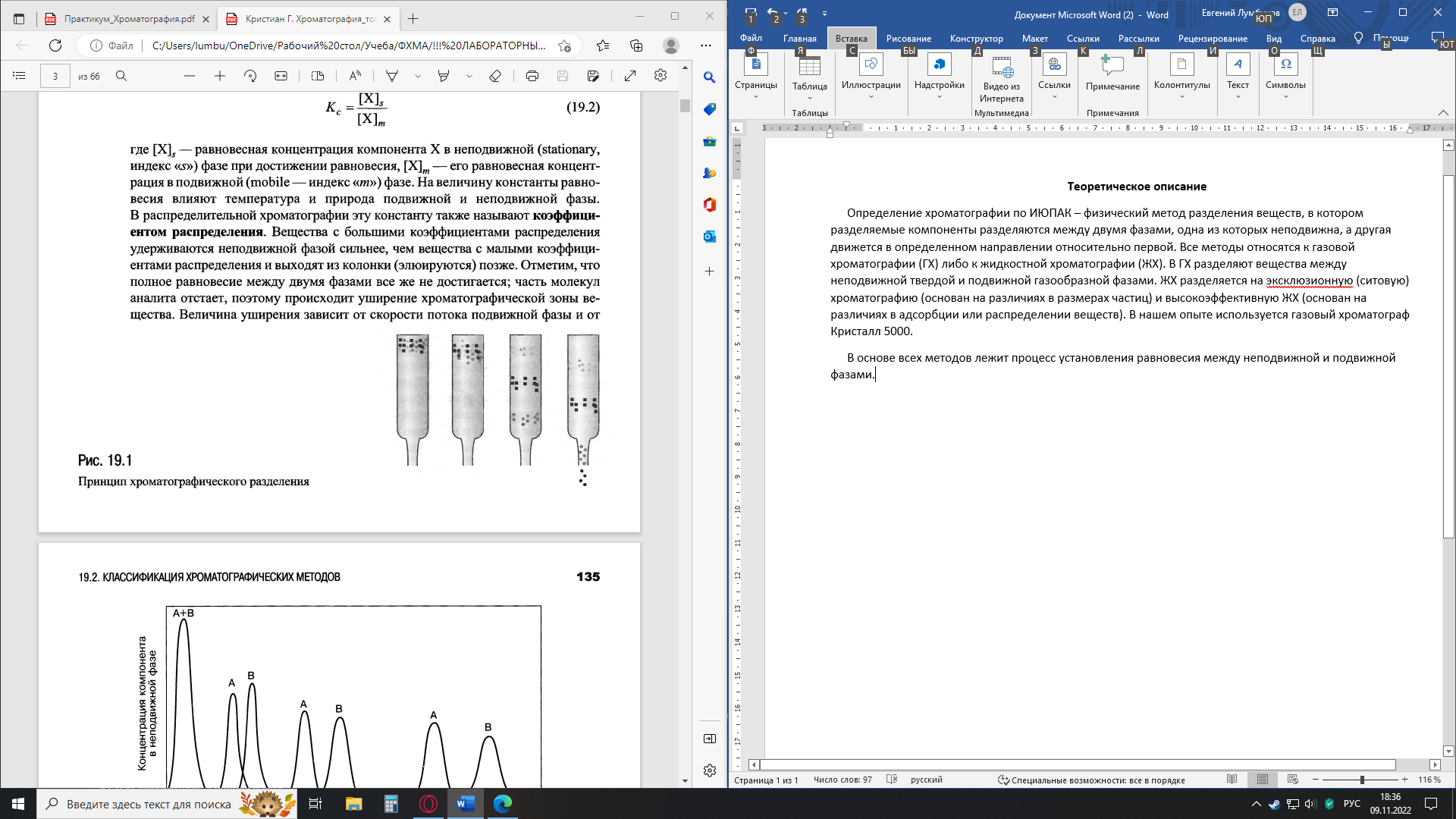 Небольшой объем образца помещают в верхнюю часть колонки, заполненной частицами неподвижной фазы (НФ). Подвижная фаза (ПФ) подается в колонку сверху и медленно по ней передвигается. Равновесие описывается константой распределения Сущность метода ГХ состоит в следующем. Анализируемая смесь (обычно — раствор) летучих компонентов переводится в парообразное состояние и смешивается с потоком инертного газа-носителя, образуя с ним ПФ. Эта смесь проталкивается далее новой порцией непрерывно подаваемого газа-носителя и попадает в хроматографическую колонку, заполненную неподвижной (стационарной) твердой или жидкой фазой — НФ. Разделяемые компоненты распределяются между ПФ и НФ в соответствии с их коэффициентами распределения. Поток газа-носителя увлекает с собой разделяемую парообразную смесь вдоль хроматографической колонки, так что процессы сорбция-десорбция разделяемых компонентов повторяется многократно, причем каждый раз в системе устанавливается динамическое равновесие разделяемых веществ между ПФ и НФ. Эти многократные переходы разделяемых веществ из ПФ в НФ и обратно совершаются по всей длине хроматографической колонки до тех пор, пока пары разделяемых веществ не покинут колонку вместе с газом-носителем. Поскольку сродство различных разделяемых веществ к НФ различно, то в процессе сорбционных—десорбционных переходов компоненты перемещаются вдоль колонки с разной скоростью. Чем больше коэффициент распределения вещества, тем дольше оно находится в НФ, тем позже покидает хроматографическую колонку. В конце концов из хроматографической колонки вместе с газом-носителем выходят зоны (объемы) парообразных хроматографируемых веществ, разделенных полностью или частично. Пары разделенных компонентов вместе с газом-носителем поступают в детектор хроматографа, генерирующий электрический сигнал — тем больший, чем выше концентрация компонента в парогазовой смеси. Электрический сигнал усиливается и фиксируется регистратором хроматографа в виде хроматограммы, записываемой на диаграммной ленте или на мониторе компьютера (если таковым снабжен хроматограф). Эти хроматограммы и используются для качественной и количественной обработки результатов анализа разделяемой смеси компонентов. Блок-схема газового хроматографа:  Блок подготовки и регулировки расхода газа. Блок регулировки расхода газа должен обеспечивать подачу газа-носителя в колонку с определенной скоростью. Для обеспечения воспроизводимых параметров разделения и оптимальных характеристик большинства типов детекторов необходима точная установка давления и расхода газа-носителя в колонке, а также поддержание их на заданном уровне с минимальной погрешностью. Типичный диапазон объемных скоростей газа-носителя при работе с насадочными колонками составляет 10–50 мл/мин, с капиллярными – 0,3–5,0 мл/мин. В большинстве неавтоматизированных газовых хроматографов давление на входе в колонку измеряют образцовым манометром. Для измерения расхода газа-носителя применяют пенные измерители, ротаметры и электрические методы (с помощью датчиков). Дозаторы, испарители. Дозирование – разделение – детектирование – в этой последовательности стадий любого хроматографического процесса ввод пробы во многом определяет конечный результат всего процесса в целом. Выбор способа дозирования зависит от природы образца, решаемых аналитических задач, типа и режима работы колонок. Любая система ввода пробы должна удовлетворять следующим требованиям: сохранять неизменность количественного и качественного состава смеси до и после дозирования (внутренняя поверхность дозатора не должна обладать адсорбционной и каталитической способностью к компонентам анализируемой смеси); обеспечивать максимальную точность и воспроизводимость величины и состава пробы; вносить минимальное размывание хроматографических пиков; идеальным считается случай, когда вся проба из дозатора, попадая в хроматографическую колонку, умещается на первой теоретической тарелке. Для ввода жидких проб чаще всего используют микрошприцы, которые позволяют отмерять объем от долей микролитров (мкл) до нескольких десятков микролитров. Проба вводится микрошприцем через самоуплотняющуюся мембрану из термостабильного силиконового эластомера либо в испаритель хроматографа, либо непосредственно в колонку. Широко распространен автоматический метод ввода газообразных проб при помощи вращающейся шайбы или газовых кранов. Конструкция кранов-дозаторов предусматривает использование сменных дозирующих петель объемом от 0,01 до 10 мл. 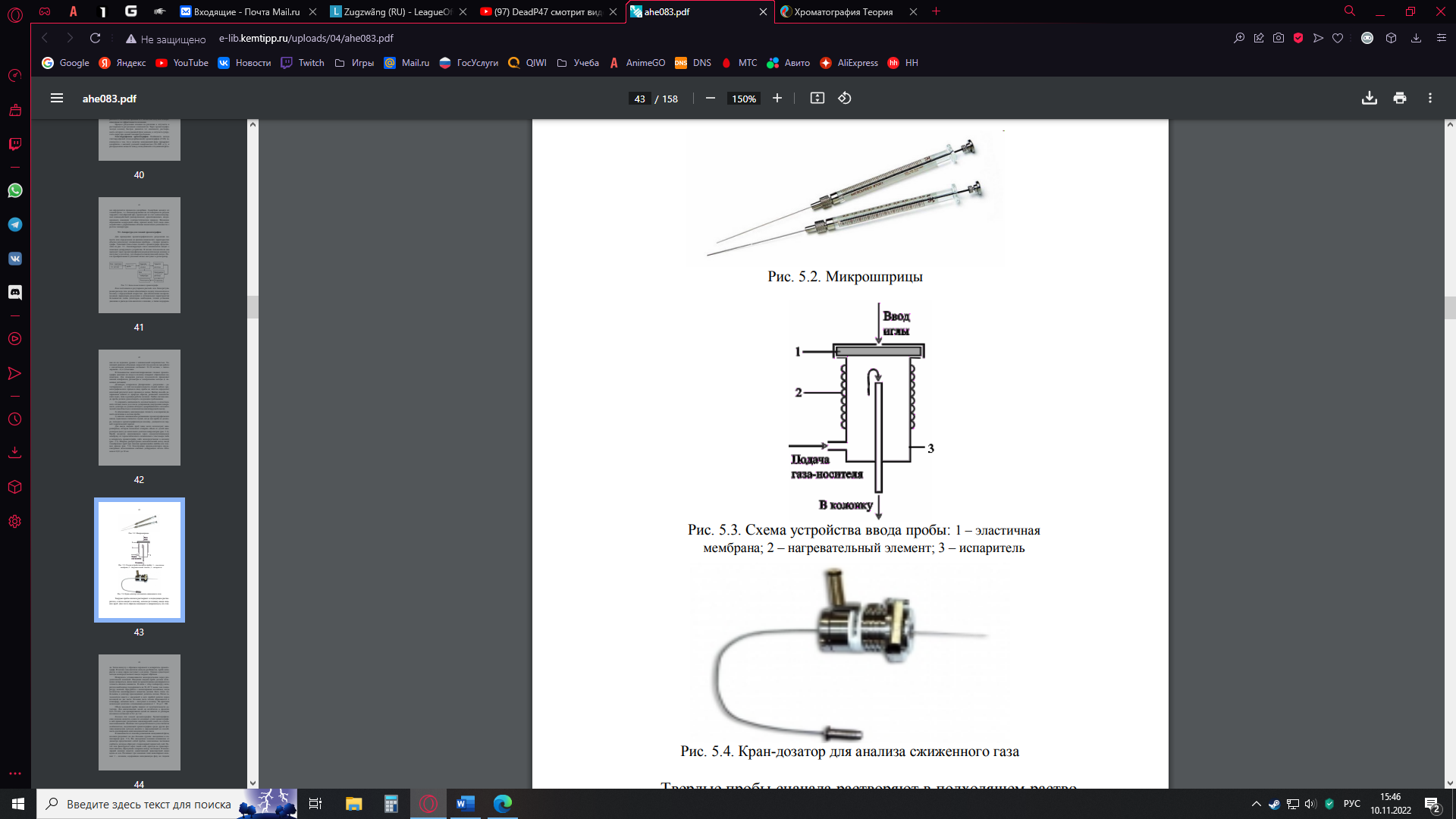 Микрошприцы Микрошприцы Схема устройства ввода пробы 1 – эластичная мембрана 2 – нагревательный элемент 3 - испаритель Кран-дозатор для анализа сжиженного газа Твердые пробы сначала растворяют в подходящем растворителе, а затем вводят в колонку, используя технику ввода жидких проб. Для этого образец помещают в микрокапсулу из стекла. Затем капсулу с образцом переносят в испаритель хроматографа. В потоке газа-носителя капсула разбивается, проба испаряется и виде паров поступает в колонку. Однако существуют методы непосредственного ввода твердых образцов. Испаритель устанавливается непосредственно перед разделительной колонкой. Вводимая жидкая проба должна мгновенно испариться, иначе пики на хроматограмме расширяются и точность анализа снижается. В связи с этим температуру испарителя необходимо поддерживать на 50–60 ˚С выше, чем температуру колонки. При работе с капиллярными колонками, когда количество анализируемого вещества должно быть очень небольшим, к дозатору присоединяют делитель потока. Поток газа-носителя вместе с введенной в него пробой делится перед колонкой на две части: большая часть потока сбрасывается в атмосферу, меньшая часть – поступает в колонку. На практике используют делители с отношением деления от 1:10 до 1:200. Объем вводимой пробы зависит от чувствительности детектора. Для аналитических целей он колеблется в пределах 0,01–10 мкл, для препаративных целей он зависит от размеров колонки и составляет от 0,1 до 1 кг. Колонки для газовой хроматографии. Хроматографическая колонка является одним из основных узлов хроматографа: в ней происходит разделение анализируемой смеси на отдельные компоненты. Наличие этого разделительного узла считается особенностью, выделяющей хроматографию среди других физико-химических методов анализа и определяющей ее способность анализировать многокомпонентные смеси. В зависимости от способа размещения неподвижной фазы, колонки разделяют на две большие группы: насадочные и капиллярные. Все насадочные колонки независимо от диаметра представляют собой трубки, заполненные частицами сорбента, которые образуют стационарный зернистый слой. Поток газа фильтруется через такой слой, проходя по транспортным каналам, образуемым зазорами между частицами. В капиллярной колонке имеется единственный транспортный канал вдоль ее оси. Различают три основных типа капиллярных колонок: 1 – колонки, содержащие неподвижную фазу на гладких стенках колонки; 2 – колонки, содержащие на стенках слой пористого сорбента; 3 – колонки, содержащие на стенках твердый носитель, пропитанный неподвижной фазой. Колонки изготовляют из коррозийно-стойкой стали, меди, стекла, латуни, полимерного материала. В последнее время капиллярные колонки изготавливают из плавленого кварца высокой чистоты, по возможности не содержащего примесей оксидов металлов. Колонки применяют различной формы: прямые, U-образные, в виде спиралей. Кварцевый капилляр, в отличие от стеклянного, вытягивается не в виде спирали, а прямым и наматывается на катушку. Размеры колонок могут быть различными в зависимости от цели анализа. Обычные насадочные колонки имеют внутренний диаметр 2–4 мм и длину 0,5–4,0 м. В микронасадочных колонках диаметр составляет 0,8–1 мм, длина – 1 м; в капиллярных диаметр равен 0,2–0,5 мм, длина колеблется от нескольких десятков до сотен метров; диаметр препаративных колонок достигает 10–100 мм и более. Термостаты. Подвижность разделяемых компонентов в колонке в большой степени зависит от температуры. Чтобы элюирование длилось определенное время, в колонке необходимо постоянно поддерживать выбранную температуру в очень узком интервале (±0,1 ˚С). Современные термостаты позволяют поддерживать температуру с такой степенью точности. Хроматографические термостаты снабжены воздушным нагревателем и вентилятором. Разделение можно проводить в изотермическом режиме и в режиме программирования температуры. При изотермическом режиме температура колонок поддерживается постоянной в ходе всего процесса разделения. Этот режим является оптимальным для разделения смеси веществ, температуры кипения которых находятся в достаточно узком интервале. Когда же они различаются значительно (более чем на 100 °С), разделение осложняется. Если поднять температуру колонки слишком высоко, то наиболее летучие компоненты выйдут слишком быстро и плохо разделятся. При более же низких температурах, во-первых, значительно увеличится общее время анализа, во-вторых, наименее летучие компоненты будут находиться в колонке столь долго, что их пики окажутся очень размытыми. Эти трудности можно преодолеть, используя режим программирования температуры. Повышение температуры в ходе анализа способствует более раннему выходу каждого последующего высококипящего компонента в виде узких пиков, и длительность анализа сокращается. Таким путем удалось разделить смесь жиров органических кислот, полученных из хлебной закваски, от эфира муравьиной кислоты до эфира пальмитиновой кислоты, молекула которой содержит 16 углеродных атомов. Детекторы. Система детектирования хроматографа – устройство, измеряющее и регистрирующее результаты хроматографического анализа. Детектор непрерывно измеряет концентрацию компонентов на выходе из хроматографической колонки и преобразует концентрацию в электрический сигнал, который регистрируется. Для детектирования используются самые различные физические и химические свойства веществ, содержащихся в газеносителе в виде паров. Необходимым свойством любого устройства, используемого в качестве детектора, является пропорциональная зависимость между сигналом и концентрацией определяемого вещества (линейность детектора), поскольку содержание последнего в анализируемой пробе может варьироваться от нескольких миллиграммов до нескольких пикограммов (10–12 г). Чувствительность детектора должна быть высокой, а минимально определяемое количество вещества (предел обнаружения) – как можно меньшим. Идеальная детектирующая система также должна давать качественную информацию о соединении, позволяющую охарактеризовать его свойства и осуществить идентификацию. Дрейф нулевой линии и фоновые шумы должны быть минимальными. Чувствительность концентрационных детекторов определяется как отклик детектора в расчете на содержание вещества в единице объема (мВ·см3/мг); чувствительность потокового детектора – в расчете на массу вещества, поступающую в детектор в единицу времени (мВ·с/мг). Минимально определяемой концентрации (нижнему пределу детектирования) должен соответствовать сигнал, в два раза превышающий уровень шума (флуктуации). Ни один детектор в отдельности не обладает сразу всеми перечисленными свойствами хотя бы потому, что некоторые из них являются взаимоисключающими. Так, например, детектор должен быть либо селективным, либо универсальным. Поэтому детектор выбирают, исходя из поставленной задачи. Обычно достаточно одного детектора, однако в особых случаях их может быть два или несколько разных типов. В сочетании с хроматографией начинают широко применяться ряд нетрадиционных приборов и методов физико-химического анализа: масс-спектрометры, атомно-абсорбционные спектрометры, ИК и УФ-спектрофотометры. Системы регистрации и обработки хроматографического сигнала. Электрический сигнал детектора непосредственно или через систему усиления поступает на регистрирующий прибор. Усилитель должен обеспечивать получение электрического сигнала, пропорционального содержанию определяемого вещества в газе-носителе, выходящем из колонки. Большинство современных хроматографов снабжены электронной системой обработки информации, измерения площади и времени удерживания пиков, расчетов количественного состава смеси. Рассмотренная система типична для обычного газового хроматографа, используемого в количественном анализе. Газовый хроматограф может иметь и гораздо более сложную схему, содержащую несколько колонок и детекторов, компьютер с памятью на магнитных дисках и в некоторых случаях автоматическое устройство для подготовки пробы. Большинство фирм, выпускающих газохроматографическую аппаратуру, обычно включают в состав прибора не более 4–6 детекторов.  Газовый хроматограф «Кристалл 5000» предназначен для анализа газообразных и жидких веществ с температурой кипения до 300 °С. Газовый хроматограф «Кристалл 5000» предназначен для анализа газообразных и жидких веществ с температурой кипения до 300 °С. Общий вид хроматографа: Экспериментальная часть Цель работы: изучение принципа действия газового хроматографа «Кристалл 5000», определение зависимости концентрации метана и ацетонитрила от отношения прощади пика одного вещества и суммы площадей пиков. Оборудование: хроматограф «Кристалл 5000», 3 колбы на 25 мл, 3 пипетки на 5 мл, микрошприц, р-р ацетона, фильтровальная бумага. Заданы 3 раствора по 5г каждый метан-ацетонитрил с концентрациями 0,05-0,95, 0,2-0,8 и 0,6-0,4 соответственно. Рассчитываем массы веществ в растворе и их объемы при ρMeth=0,792 г/мл и ρAN=0,786 г/мл.
Запускаем хроматограф: Проверяем подключение к сети и компьютеру Подаем газ в прибор, убедившись по манометрам, что выходного давления в баллоне достаточно для работы (>2 МПа), а входное давление находится в рабочем диапазоне (от 0,4 до 0,5 МПа) Включаем все приборы и запускаем программу Задаем нужные нам параметры и ждем подготовки 30 минут (температурный режим – 80˚С – ДТП, термостатированная колонка - 90˚С, колонка капиллярная 60 м с сечением 0,32 мм, фаза – полиэтиленгликоль, метод калибровки – метод внутренней нормализации) По рассчитанным объемам готовим растворы, определяя на аналитических весах массы веществ
Выждав подготовку, проводим анализ чистых компонентов для определения пиков в будущем (перед забором пробы, промываем микрошприц ацетоном 5 раз, затем, набираем и выплескиваем на фильтровальную бумагу анализируемый раствор 5 раз и только затем набираем 1 мкл, удостоверившись, что в микрошприце нет пузырьков воздуха, после чего, перед следующим забором промываем также ацетоном) τвыхода, Meth = 4,42 минуты, SMeth = 44427, τвыхода, AN = 5,71 минуты, SAN = 45826 Анализируем приготовленные растворы по 2 раза для уменьшения погрешности и записываем площади пиков в таблицу
Проверяем результаты на ошибки, путем сравнения отношений площадей пиков Meth к AN, грубые ошибки отсутствуют Рассчитываем концентрации веществ через массы, полученные на весах
Строим график зависимости концентраций от отношения площади пика к сумме площадей пиков 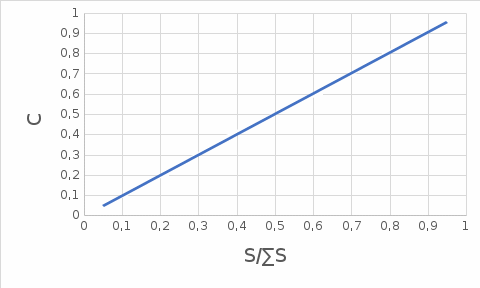 Делаем вывод, что доля площади пика линейно зависит от концентрации вещества в растворе Литература Кристиан Г. «Аналическая химия, том 2» Заворотный В.Л., Калачева Н.А., Зайцев Н.К. «Учебное пособие по курсу Аналитическая химия (Хроматография)» В.П. Гуськова, Л.С. Сизова «Хроматографические методы разделения и анализа» |