СИНДРОМ «ВЯЛЫЙ РЕБЁНОК». УЧЕБНО-МЕТОДИЧЕСКОЕ ПОСОБИЕ.. Учебнометодическое пособие для студентов педиатрических факультетов, интернов, ординаторов и врачей педиатров. Синдром вялый ребёнок
 Скачать 178.61 Kb. Скачать 178.61 Kb.
|

|
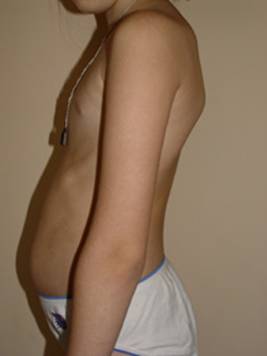
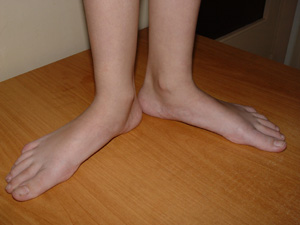
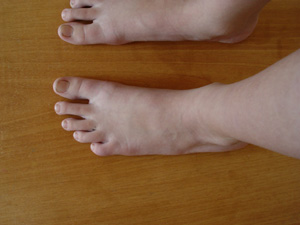
МиНИСТЕРСВО ЗДРАВООХРАНЕНИЯ И СОЦИАЛЬНОГО РАЗВИТИЯ РФ Кафедра педиатрии Учебно-методическое пособие для студентов педиатрических факультетов, интернов, ординаторов и врачей педиатров. Синдром «вялый ребёнок» 1. Введение. Термин «вялый ребёнок» был предложен в 1958 году Greenfield J.G. и соавторами для обозначения врождённой мышечной гипотонии независимо от её генеза. В современной медицинской литературе это термин широко используется как синдром «вялый ребенок». Известно около 80-ти заболеваний, проявляющихся в раннем детском возрасте симптомокомплексом, в основе которого лежит диффузная мышечная гипотония, поэтому их дифференциальная диагностика затруднена. Целесообразность введения этого термина была обусловлена тем, что врождённая амиотония, описанная в 1900 году Oppenheim H., как самостоятельная нозологическая форма отсутствует. В частности, Oppenheim H. предполагал, что в основе амиотонии лежит врождённое недоразвитие мышц. К его мнению присоединились в 1911 году Архангельский В.Г. и Абрикосов А.И. К настоящему времени широкое использование в медицине морфологических и, особенно, биохимических методов исследования позволило выявить связь врождённой мышечной амиотонии с группой врождённых, медленно прогрессирующих миопатий, спинальной амиотрофией Верднига-Гоффмана, патологией щитовидной железы, перинатальной патологией нервной системы, наследственными заболеваниями обмена веществ (аминоацидурии, гликогенозы, липидозы и т.д.), хромосомными синдромами, наследственными болезнями срединительной ткани и другими заболеваниями, проявляющимися в периоде новорожденности мышечной гипотонией (Бадалян Л.О., Скворцов И.А., 1979, 1986; Dubovitz V., 1980,1986 ; Royden J.H., 1990 ; Crawford A.D., 1992). 2. Этиология и патогенез. По данным ряда авторов синдром «вялый ребёнок» рассматривается как полиэтиологическая патология. Клинические проявления синдрома неспецифичны, а его течение и исход различны в каждом конкретном случае. В основе снижения мышечного тонуса лежат общие патофизиологические механизмы. В регуляции мышечного тонуса принимают участие структуры ствола головного мозга, стриопаллидарная система, мозжечок, но в конечном итоге реализация тонических реакций осуществляется с деятельностью образований сегментарного рефлекторного аппарата: альфа – и гамма – нейронами передних рогов спинного мозга, двигательными и чувствительными волокнами нервных стволов, афферентами нервно-мышечных веретён (Granit R., 1973; Birkmayer W., 1974). Клиническая картина синдрома «вялый ребёнок» весьма характерна: резкая мышечная гипотония, достигающая в ряде случаев степени полной атонии, снижение или полное отсутствие активных движений, безусловных рефлексов. Общая мышечная слабость сопровождается у ребенка затруднением сосания и дыхательной недостаточностью. Отсутствуют характерные установки позы, приподнятые конечности падают, как плеть. Вследствие мышечной гипотонии отмечается резкое переразгибание суставов, их разболтанность. Таким образом, мышечная гипотония является частым случаем патологии в периоде новорожденности и указывает на тяжёлые нарушения у ребёнка. Определение причин этих нарушений является для врача особенно трудной задачей. Точная диагностика мышечной гипотонии важна также для адекватной терапии и медико-генетического консультирования в отягощённых семьях. На приведенной схеме представлены патогенетические механизмы наследственных заболеваний, проявляющихся синдромом «вялый ребенок». 3. Клинико-генетическая характеристика ряда наследственных заболеваний, протекающих с диффузной мышечной гипотонией. Изучение причин мышечной гипотонии у детей раннего возраста и анализ данных литературы позволяет выделить ряд наиболее характерных наследственных заболеваний и синдромов, которые могут сопровождаться врождённой амиотонией (Бадалян Л.О., Скворцов И.А., 1986; Royden J.H., 1990). Причем следует заметить, что клиническая значимость синдрома мышечной гипотонии требует от врача правильной интерпретации и осторожности при оценке степени его тяжести. 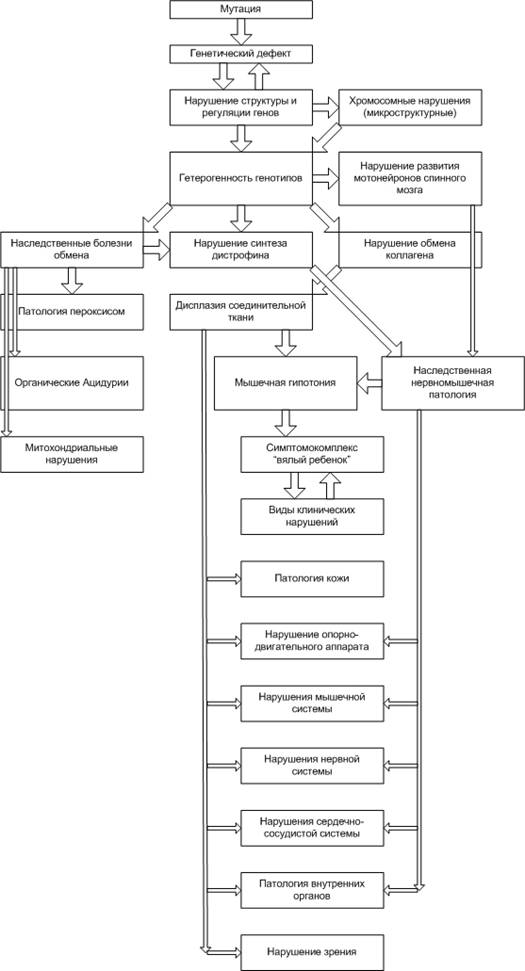 3.1. Синдром Элерса–Данлоса. Среди наследственных заболеваний соединительной ткани мышечная гипотония встречается при одном из наиболее частых заболеваний этой группы – синдроме Элерса-Данло, представляющем собой генетически гетерогенное заболевание, поражающее суставы и кожу и различающееся по типам наследования, клиническим особенностям, а также по первичному молекулярному и биохимическому дефекту. Частота синдрома составляет 1 : 100000 новорожденных (Блинникова О.Е., 1985). В основе патогенеза мышечной гипотонии при наследственных заболеваниях соединительной ткани лежит первичная патология коллагена. В этом случае дефект механизма синтеза и секреции коллагена, неправильное расположение коллагена вне клетки ведут к клиническому проявлению заболевания. Синдром Элерса-Данло – болезнь, затрагивающая многие системы организма. В начале ХХ столетия были сделаны попытки описать необычную семейную патологию соединительной ткани, характеризующуюся гипермобильностью суставов, сверхрастяжимостью, хрупкостью кожи и склонностью к гематомам и шрамам. Локализация патологического гена была установлена в последнее десятилетие, но далеко не для всех форм синдрома. Kuivaniemi H. и соавт. (1989) сообщили о четырёх точковых мутациях в гене проколлагена типа II , локализованном в сегментах 2q24.3–q31 и вызывающем тяжёлые и промежуточные формы синдрома IV типа. Молекулярные механизмы патологии соединительной ткани также выявлены не для всех форм синдрома. Имеются сведения о нарушении межмолекулярных поперечных связей и необычном строении коллагеновых фибрилл у некоторых больных с I типом синдрома (Вельтищев Ю.Е., Ананенко А.А., 1985). Клинический диагноз синдрома Элерса–Данлоса основан на следующих симптомах: гипермобильность суставов, сверхрастяжимость кожи, аномальное рубцевание, склонность к кровоизлияниям (см. Клинический пример). При различных типах синдрома внешние признаки заболевания чрезвычайно сходны между собой и практически нет критериев для их клинической дифференцировки. Характерны гиперрастяжимая, тонкая и лекго ранимая кожа, её слабая фиксация с подлежащими тканями. Кожу можно приподнять на 1,5–2 см в тех местах, где это невозможно у здорового человека, например, на кончике носа или ушных раковинах. При минимальном травмировании кожи легко возникают разрывы, крайне медленно заживающие, с образованием атрофических рубцов и тонкой лоснящейся морщинистой поверхностью, так называемые «папиросные рубцы». Слизистые оболочки при синдроме Элерса–Данлоса характеризуются аналогичными с кожей свойствами. Весьма характерным для синдрома является поражение мышечной системы в виде её диффузной гипотонии и слабого развития. Тяжёлая мышечная гипотония нередко является причиной ошибочной диагностики врождённой амиотонии, а иногда может имитировать нервно- мышечные заболевания. Следующий типичный признак синдрома - это гиперподвижность суставов, в результате чего больные могут выполнять сложные акробатические упражнения без предварительной тренировки. Манифестация этой способности обычно проявляется в конце первого - начале второго года жизни, когда ребёнок начинает самостоятельно ходить. Следствием гиперподвижности могут быть повторные вывихи и подвывихи суставов, иногда осложняющиеся выпотом в суставную сумку и остеоартритом. Патология опорно-двигательного аппарата характеризуется кифосколиозом, воронкообразной деформацией грудной клетки, высоким сводом стопы, косолапостью плоскостопием. Некоторые из перечисленных симптомов достаточно ярко выражены у девочки с верифицированным синдромом Элерса-Данло (см. фото.)
Довольно часто синдром сопровождается геморрагическим диатезом. У большинства больных легко возникают экхимозы, кровоподтёки, гематомы различной локализации. Типичны кровотечения из дёсен, носа, матки, желудочно-кишечного тракта, продолжительная необъяснимая гематурия. Нередко встречаются изменения глаз: птоз, косоглазие, голубые склеры, эктопия хрусталиков, миопия. В ряде случаев у пробандов с синдромом Элерса–Данло обнаруживается частичное отсутствие зубов, их неправильное формирование, аномальное расположение и уменьшение в размерах (Блинникова О.Е., 1985). В литературе описаны разрывы сосудов крупного и мелкого калибра, множественные аневризмы сосудов головного мозга и аорты, артерио-венозные фистулы (McKusick V.A.,1972). Лёгочная патология проявляется эмфиземой, спонтанным пневмотораксом, трахеобронхомегалией (Cupo L., 1981). Описаны также больные с выпадением прямой кишки, спонтанными перфорациями кишечника, мегаколоном, спланхоптозом, дивертикулами желудочно-кишечного тракта. У таких больных легко возникают грыжи, обусловленные снижением механической прочности фасций (McKusick V.A., 1972). Дети с синдромом Элерса-Данло часто рождаются недоношенными вследствие хрупкости околоплодных оболочек, нередко имеют врождённые вывихи тазобедренных и плечевых суставов. Имеются указания на уменьшенную массу тела у новорожденного (McKusick V.A., 1972). У матерей детей с синдромом Элерса-Данло нередко возникают акушерские и хирургические проблемы, связанные с преждевременным излитием околоплодных вод у беременных, послеродовыми кровотечениями, расхождением лобкового симфиза и наложенных швов, плохим их заживлением, выпадением матки. 3.2. Прогрессирующая мышечная дистрофия. Слабость и «похудание» мышц, ограничение или полное отсутствие активных движений в конечностях составляют клиническую картину заболеваний с поражением нервно-мышечной системы. В их числе прогрессирующая мышечная дистрофия (ПМД) Дюшенна. Первое наблюдение псевдогипертрофической миопатии принадлежит Meryon E. (1852). В 1861 году Duchenne G.B.A. описал больного с псевдогипертрофическим мышечным параличом, обратив внимание на необычное сочетание: увеличение икроножных мышц и прогрессирующую мышечную слабость. При дальнейшем изучении дюшенновской миодистрофии выяснилось, что помимо злокачественного варианта существует вариант доброкачественного течения болезни (Becker P.E., 1955). Миодистрофия Дюшенна-Бекера наследуется по рецессивному, сцепленному с Х-хромосомой типу. Около 1/3 случаев являются результатом новых мутаций (Saito K. и соавт., 1992). Болезнь относится к классу первичных миодистрофий, при которых течение патологического процесса в мышцах обусловлено нарушением обмена веществ в них (Гринио Л.П., 1997, 1998; Агафонов Б.В., 1997). Мутации, приводящие к полному нарушению синтеза дистрофина, проявляются клиническим фенотипом ПМД Дюшенна. При отсутствии синтеза белка-дистрофина фенотипические проявления соответствуют ПМД Дюшенна. При снижении количества и изменениях размеров молекулы дистрофина фенотипические проявления соответствуют ПМД Бекера, отличающейся более поздним дебютом и доброкачественным течением (Saito K. и соавт., 1992). На современном этапе клинических исследований выделены две формы дюшенновской миодистрофии: форма Дюшенна у мальчиков и форма Беккера у мужчин. В первом случае клинические признаки более чётко выражены, чем во втором, и различия заключается в разных темпах течения патологического процесса в мышцах и, следовательно, продолжительности жизни больных. Клиническая картина миодистрофии Дюшенна хорошо изучена. Заболевание проявляется в возрасте 2-5 лет. Ранними признаками являются : двигательная неловкость, неустойчивость, частые падения. Нередко родители отмечают двигательную пассивность детей, нежелание ходить. При физической нагрузке быстро наступает утомляемость. При подъёме по лестнице дети часто останавливаются, опираются на перила. Походка приобретает «утиный» характер (Вельтищев Ю.Е., Тёмин П.А., 1998). Одним из наиболее характерных симптомов являются псевдогипертрофии различных групп мышц: икроножных, дельтовидных, ягодичных, иногда прямых мышц живота, бёдер. По мере прогрессирования заболевания псевдогипертрофии имеют тенденцию к уменьшению. Мышечные атрофии первоначально локализуются в мышцах тазового пояса и проксимальных отделах нижних конечностей, а в последующем распространяются в восходящем направлении на плечевой пояс, мышцы спины, проксимальные отделы верхних конечностей. В ранней стадии заболевания выявляется снижение или выпадение коленных рефлексов, затем снижаются и выпадают ахилловы, бицепитальные и трицепитальные рефлексы. Как сказано выше, течение ПМД Бекера более доброкачественное, чем ПМД Дюшенна. В типичных случаях заболевание возникает в возрасте 10-20 лет, иногда несколько раньше или позже (Gomi G.P. и соавт., 1994). Многие больные до 40-60 лет сохраняют способность к самостоятельному передвижению. Двигательные нарушения при форме Бекера прогрессируют не так быстро. Длительное повышение нагрузки на миокард ведёт к развитию дилятационной кардиомиопатии и митральной регургитации. Причиной смерти больных с ПМД Бекера являются сердечно- сосудистые осложнения. В настоящее время считается, что с генетической точки зрения ПМД Дюшенна и ПМД Бекера являются результатом мутации в одном и том же гене, а с клинической точки зрения – это две разные формы: злокачественная и доброкачественная соответственно. 3.3. Спинальная мышечная атрофия Верднига-Гоффманна. Одним из заболеваний, проявляющихся синдромом «вялый ребёнок», является спинальная мышечная атрофия Верднига–Гоффманна, причиной которого является дегенерация клеток передних рогов спинного мозга. Это заболевание впервые было описано у детей Werdnig G. и Hoffmann J. в 1891-1892 г.г. В 1956 году Кугельберг и Веландер выделили также форму спинальной мышечной атрофии с более поздним началом и относительно доброкачественным течением. В настоящее время выделено 3 типа спинальной мышечной атрофии. Основанием для такого разделения служат: различное время манифестации болезни, различный характер течения болезни и различная степень выраженности (экспрессивность) клинических проявлений. Выделяют: I тип - болезнь Верднига-Гоффманна, II тип – промежуточный тип болезни и III тип – болезнь Кугельберга - Веландера. Спинальные мышечные атрофии детского возраста наследуются по аутосомно-рецессивному типу и проявляются у гомозигот. Гетерозиготы фенотипически не отличаются от здоровых лиц. При мышечной атрофии I типа с момента рождения больного ребёнка наблюдается синдром «вялый ребёнок» в виде генерализованной мышечной гипотонии, слабости мышц туловища и преимущественно мышечных групп проксимальных отделов конечностей. В первые месяцы жизни возникают атрофии и фасцикулярные подёргивания в мышцах спины, туловища, проксимальных отделах конечностей. Иногда наблюдаются бульбарные расстройства: слабый крик, вялое сосание, дисфагия, фибрилляции мышц языка. Грудная клетка уплощена вследствие слабости межрёберных мышц. Также в первые месяцы жизни у большинства больных отмечаются частые аспирации, респираторные инфекции, нередко пневмония. С возрастом мышечная слабость прогрессирует, исчезают приобретенные двигательные навыки, и ребёнок, лёжа на спине, приобретает форму «распластанной лягушки». Внешне больной сохраняет облик здорового ребёнка, так как мышечная атрофия замаскирована выраженной подкожной основой, показывающей наощупь ощущение плотности. Характерным является хорошее психическое развитие, несмотря на обездвиженность ребенка. Течение заболевания быстро прогрессирующее, с летальным исходом к 1-1,5 годам жизни. Наряду с «классической» спинальной мышечной атрофией I типа, описан атипичный вариант болезни, характеризующийся респираторным дистресс-синдромом (вследствие пареза диафрагмы), прогрессирующей мышечной слабостью и атрофиями дистальных отделов конечностей, дисфагией, фибрилляциями языка. Летальный исход заболевания наблюдается на первом году жизни (Novell G. и соавт., 1994). II тип промежуточный спинальной мышечной атрофии детского возраста дебютирует в течение 18-ти месяцев жизни. Первые признаки заболевания проявляются на 8-14 месяце в виде генерализованной мышечной слабости и гипотонии. С момента манифестации заболевания отмечается задержка моторного развития ребенка. Только 25% больных детей могут самостоятельно сидеть, а в редких случаях могут стоять с поддержкой. Течение заболевания неблагоприятное, в возрасте 1-4 года наблюдается летальный исход (часто от пневмонии) в связи с поражением дыхательной мускулатуры. Особенностью течения спинальной мышечной атрофии III типа является ее относительная доброкачественность. Клинические признаки появляются после 18-ти месяцев жизни; наиболее выражены к 2-5 годам. В течение многих лет болезнь не приводит к глубокой инвалидности. В 1994 году Parano E. и соавт. описали для Ш типа заболевания наличие (наряду с мышечными атрофиями) псевдогипертрофий икроножных, ягодичных и дельтовидных мышц). 3.4. Наследственные заболевания из группы пероксисомных болезней. К группе заболеваний, проявляющихся синдромом «вялый ребенок» на первом году жизни, относятся заболевания из группы пероксисомных болезней. В их числе: атипичная и точечная хондродисплазия, дефицит бифункционального белка, инфантильная болезнь Рефсума, синдром Цельвегера, варианты синдрома Цельвегера, пипеколовая ацидемия и тригидроксихолестанемия. Основными симптомами данной группы заболеваний являются: гипотония, ареактивность, судороги, затруднения при кормлении, гепатомегалия, желтуха, нарушения пищеварения, гипохолестеринемия, дефицит витамина Е, черепно-лицевой дизморфизм (плоское, одутловатое лицо с опухшими веками, высокий лоб, сглаженные надбровные дуги, гипертелоризм, эпикант, монголоидный разрез глаз, нередко нистагм, катаракта или глаукома, микрогнатия и брахицефалия, большие роднички, высокое арковидное небо, низко расположенные ушные раковины), а также скелетные аномалии (ассиметричное укорочение конечностей в связи с точечной кальцификацией эпифизов, контрактуры крупных суставов, ранний сколиоз, нанизм). При выявлении выше перечисленных симптомов необходимо направление больного ребёнка в генетико-биохимическое учреждение для определения нарушенной функции пероксисом. 3.5. Наследственные болезни обмена. Многие наследственные болезни обмена (НБО) проявляются уже в периоде новорожденности, но не всегда распознаются, так как рассматриваются врачами как последствия интранатальной гипоксии, внутричерепной родовой травмы или фетального респираторного дистресса. Отличительной особенностью НБО у новорожденных служит наличие бессимптомного периода (первые двое-трое суток после рождения). Дети, как правило, рождаются внешне здоровыми в результате нормально протекавшей беременности. Состояние ребенка ухудшается внезапно с проявлениями энцефалопатии (нейродистресс–синдром). Аномалии обмена, обусловленные ферментопатиями, по клиническим проявлениям делятся на две группы: токсического или гипоэнергетического типа. Энцефалопатии токсического типа связаны с накоплением токсических продуктов, синтезирующихся в метаболической цепи до ферментативного блока. Ими могут быть аминокислоты, органические кислоты, аммиак, галактозо-1-фосфат и др. Ранними клиническими проявлениями являются: отказ ребенка от груди, рвота, дегидратация, судороги, летаргия, кома, остановки дыхания, мышечная и почечная недостаточность, гипертонус и клонус мышц, ацидоз, кетоз, гипераммониемия. Энцефалопатии гипоэнергетического типа обусловлены либо истощением запасов энергетических субстратов, либо невозможностью их использования организмом вследствие метаболического блока. Это наблюдается при ферментопатиях глюконеогенеза, гликолиза, окисления жирных кислот, недостаточности митохондриальных дегидрогеназ. При этих энцефалопатиях развивается такая же клиническая сиптоматика, как и при токсических энцефалопатиях, но с более выраженной мышечной гипотонией, гипо- или арефлексией, кардиомиопатией, сосудистой недостаточностью и коллапсом, иногда – внезапной смертью (синдром «внезапной смерти»). Время проявления первых клинических симптомов, степень их выраженности и темпы прогрессирования при НБО вариабельны и зависят от характера метаболического дефекта и остаточной активности дефектного фермента. Чем ниже остаточная активность фермента, тем раньше возникают метаболические расстройства и, соответственно, клинические проявления. Степень тяжести заболевания и время его проявления также зависят от токсичности накапливающихся в организме продуктов метаболизма. Выделяют 4 варианта клинического течения НБО (Журба Л.Т., Мастюкова Е.М., 1981). Из них только один вариант имеет особенности манифестации на первом году жизни и характеризуется острым течением и ранней манифестацией клинических проявлений (в первую очередь неврологических нарушений, патогенетически связанных с нейротоксикозом. В клинической картине на первый план выступают общемозговые симптомы, определяющие тяжесть состояния ребёнка. Уже с рождения или в первые дни наблюдается заторможенность, апатия, гипомимия, слабый крик, частая рвота, коматозное состояние или, наоборот, резкое беспокойство, пронзительный крик, нарушение сна, тремор, судороги. Расстройства черепно-мозговой иннервации проявляются преходящим косоглазием, анизокорией, спонтанным нистагмом, дисфагией, иногда ларингоспазмом, нарушением дыхания. Мышечный тонус резко снижен. Сухожильные рефлексы, наоборот, высокие с расширенной зоной. Часто самым первым симптомом заболевания являются судороги. Затем быстро нарастают неврологические нарушения, развивается дистрофия. Ребёнок принимает позу опистотонуса, учащаются судороги. Больные нередко погибают в ближайшие дни или недели после появления первых симптомов. Если дети выживают, то в дальнейшем у них развиваются глубокая задержка психомоторного развития, микро- или гидроцефалия, частые судорожные припадки, резкое снижение слуха и зрения. Ранняя клиническая манифестация наблюдается при следующих НБО: нарушения аминокислотного обмена: аргининемия, аргининянтарная аминоацидурия, гипервалинемия, лейциноз, тирозиноз, цитруллинемия; реже - гистидинемия, изовалериановая ацидемия и фенилкетонурия; нарушения углеводного обмена (галактоземия, гликогенозы); нарушения липидного обмена (болезнь Гоше, болезни Нормана-Вуда и Нормана- Ландинга, ганглиозидоз Gm 3); дефекты обмена витаминов (метилмалоновая ацидурия, некетоническая и кетоническая гиперглицинемия, некротизирующая энцефаломиелопатия, пиридоксин - зависимый судорожный синдром, тиамин-зависимый лактатацидоз, фолат-зависимая мегалобластическая и другие формы нарушения обмена фолиевой кислоты); нарушения обмена минеральных веществ (гипофосфатазия, синдром «кудрявых волос» Менкеса). 3.6. Хромосомные и микрохромосомные нарушения. Признаки синдрома «вялый ребёнок» и, в частности, мышечная гипотония - это частая симптоматика при хромосомных и микрохомосомных нарушениях у детей: синдромы Дауна, Ангельмана, Прадера-Вилли и др. Для такого рода нарушений также присущ ряд своих специфических проявлений, включая: 1. Клинико-генеалогические особенности (много – или маловодие, угроза прерывания беременности, самопроизвольные аборты, мёртворождения, преждевременные роды). 2. Врожденный характер заболевания и симметричность поражения. 3. Пренатальная гипотрофия. 4. Особый признак - множественные врождённые пороки развития. 5. Малая для срока гестации окружность головы. 6. Задержка психомоторного развития. 4. Причины клинического полиморфизма при синдроме «вялый ребёнок». Проблема клинического полиморфизма (разнообразие клинической картины при одном и том же заболевании у разных лиц) является центральной проблемой клинической медицины независимо от её профессиональной направленности. Феномен клинического полиморфизма наследственных болезней нервной системы у детей впервые обозначил и проанализировал ещё в 30-ые годы прошлого столетия выдающийся русский врач-невропатолог и первый отечественный врач-генетик Сергей Николаевич Давиденков. К настоящему времени медициной накоплен огромный фактический материал по феноменологии клинического полиморфизма при отдельных формах наследственных заболеваний нервной системы. Генетические причины клинического полиморфизма в широком смысле обусловлены неповторимой генотипической и фенотипической индивидуальностью человека как биологического вида (гетерогенностью генотипов). Именно гетерогенность генотипов является основой полиморфизма признаков, определяющих понятие - синдром «вялый ребёнок» и заболеваний, протекающих с диффузной мышечной гипотонией. Клиническая картина при синдроме Элерса-Данло обусловлена плейотропным действием гена и вовлечением в патологический процесс многих систем организма (полисистемный характер поражения). Наблюдаемый при этом клинический полиморфизм объясняется наличием нескольких генетических форм основного заболевания, отличающихся различиями в первичных молекулярных и биохимических дефектах, а также по типам наследования болезни и экспрессивности ее клинических проявлений (всего 10 типов). Вместе с тем внешние признаки различных генетических типов синдрома Элерса-Данло чрезвычайно сходны между собой и практически нет чётких критериев для их клинической дифференцировки. Это касается гиперрастяжимости и ранимости кожи, гиперподвижности суставов и симптомов геморрагического диатеза. Одинаковая (или сходная) клиническая картина при различных типах синдрома Элерса-Данло обусловлена феноменом генокопирования или полилокусности. При этом каждый отдельно взятый генетический тип синдрома Элерса – Данлоса есть результат мутации в отдельно взятом гене (локусе) среди множества генов, имеющих отношение к синтезу белка волокнистых элементов соединительной ткани, главным образом, коллагеновых волокон. Такие мутации, затрагивающие разные генные локусы, как бы копируют клиническую картину болезни (Мутовин Г.Р., 2001). В случаях миодистрофии Дюшенна и Бекера фенотипические различия по проявлению признаков болезни могут быть обусловлены генетическими различиями в механизмах взаимодействия двух аллельных генов или множественным аллелизмом. Полиморфизм признаков может зависеть от изменения различных участков одного и того же гена. Например, мутация внутри гена может затрагивать разные участки на всём его протяжении, причём фенотипически более выраженными будут повреждения экзонной части гена (содержит наследственную информацию) по сравнению с интронами (не содержат наследственную информацию). Таким образом, из приведённых выше примеров следует, что в рамках одной нозологической формы существуют фенотипы, обусловленные множественным аллелизмом, разными мутациями одного и того же гена, мутациями в разных генах, что и определяет особенности клинического проявления и течения патологического процесса у различных больных. Можно заключить, что клинический полиморфизм при синдроме «вялый ребёнок» обусловлен множеством генетических причин. В случае синдрома «вялый ребёнок» генетические причины клинического полиморфизма, вероятно, могут быть связаны с действием со стороны других генов: генов–модификаторов. Вместе с патологическим геном индивид наследует от родителей комбинации других генов, которые в каждом конкретном случае могут усиливать (гены–интенсификаторы) или ослаблять (гены-супрессоры) действие патологического гена. Важно отметить, что для практической медицины определение генетических (и негенетических) причин полиморфизма признаков синдрома «вялый ребёнок» открывает новые возможности для диагностики, выбора методов лечения и предупреждения заболевания, в том числе в процессе медико-генетического консультирования. При этом врач определяет прогноз течения заболевания у больного с синдромом «вялый ребёнок» и прогноз здоровья будущего потомства в отягощённой семье. 5. Методы диагностики синдрома «вялый ребёнок». 1. Клинико-генеалогический метод. Позволяет установить наследственный характер заболевания или признака, определить тип наследования и пенетрантность (вероятность проявления) патологического гена, выявить гетерозиготное носительство такого гена, проводить медико-генетическое консультирование (определить прогноз заболевания и прогноз для будущего потомства). 2. Молекулярно-генетический метод. Применяется в целях уточнения диагноза при ряде наследственных заболеваний, обусловливающих клинические проявления синдрома «вялый ребёнок» (ПМД Дюшенна и ПМД Бекера, спинальная мышечная амиотрофия Верднига-Гоффманна). Решение вопроса о проведении молекулярно-генетического исследования принимается после окончательного уточнения диагноза основного заболевания. 3. Функциональные методы исследования: 3.1. Метод электромиографии и электронейромиографии. Играют значительную роль в изучении синдрома «вялый ребёнок». Они позволяют дифференцировать преимущественное поражение мышцы, нерва или передних рогов спинного мозга (Бадалян Л.О. и соавт., 1979). Около 80% случаев синдрома «вялый ребенок» обусловлены первичным поражением ЦНС и для них не требуется применение ЭМГ и ЭНМГ, так как клинический диагноз легко распознаётся внимательным невропатологом. В остальных случаях специфическая картина не вполне очевидна, и тогда ЭМГ является полезной составной частью обследования, так как демонстрирует очевидные результаты при различных состояниях с нарушением движения ребёнка. 4. Ультразвуковые методы исследования : 4.1. Метод эхо-кардиографии. Позволяет диагностировать патологию сердечной мышцы, клапанов сердца и крупных сосудов, что достаточно часто встречается при синдроме «вялый ребёнок». 4.2. Метод ультразвукового исследования внутренних органов. Позволяет определить положение, размер, эхогенность, степень васкуляризации, наличие аномалий развития различных внутренних органов, а также состояние прилежащих к ним мягких тканей. 4. Заключение. При выявлении признаков синдрома «вялый ребенок», специфичных для перечисленных выше наследственных заболеваний, врач- педиатр (врач-неонатолог) должен направить больного ребёнка на обследование в генетико-биохимическое учреждение для уточнения диагноза и определения причин, вызвавших данный симптомокомплекс. При отсутствии дополнительной специфической симптоматики на первом году жизни ребенка необходимо динамическое наблюдение за ним с целью определения дальнейшей тактики обследования и ведения. |