Законов идеальных растворов учитывается при помощи расчетной величины активности (Г. Льюис)
 Скачать 0.69 Mb. Скачать 0.69 Mb.
|

Способы определения коэффициента активности реальных растворов неэлектролитов.В неидеальных растворах (жидких и твердых) отклонение от законов идеальных растворов учитывается при помощи расчетной величины — активности (Г. Льюис). Зависимость химического потенциала μi компонента в идеальном растворе от молярной доли хi, этого компонента в растворе выражается уравнениями: dμi= RT d ln xi ; (1) μi=μ*i+RT ln xi ; (2) μi,2=μi,1+RT ln(xi,2/xi,1) ; (3) где μ*i — химический потенциал чистого i-го компонента (хi= 1); μ*i зависит от температуры, давления и природы вещества; μi,1 и μi,2 — значения μi в 1-м и 2-м состояниях. Для вычисления химического потенциала i-го компонента в неидеальном растворе в (1) — (3) вместо концентрации (молярной доли) подставляется активность ai i-го компонента: dμi= RT d ln ai ; (4) μi=μ0i+RT ln ai ; (5) μi,2=μi,1+RT ln(ai,1/ai,2) ; (6) где μ0i — стандартный химический потенциал. В стандартном состоянии активность принимают равной единице, a0i = 1. Активностью ¡-го компонента раствора называется величина, которую нужно подставить в выражение для химического потенциала компонента в идеальном растворе, чтобы получить действительное значение химического потенциала ¡-го компонента в неидеальном растворе. Коэффициентом активности называется отношение активности компонента в растворе к его концентрации. Равновесные свойства неидеальных растворов зависят от активности растворителя или растворенного вещества. Рассмотрим несколько способов определения активности или коэффициента активности растворителя и растворенного вещества в неидеальных растворах. Активность зависит от выбора стандартного состояния. Определение активности летучего растворенного вещества по парциальному давлению его пара. По парциальному давлению. Используя закон Рауля  p1=p01 a1 p1=p01 γ1x1 p1=p01 a1 p1=p01 γ1x1γ1=p1/ p01x1 Активность используется только для растворов, не для газов! p2=k*a2 p2=k γ2x2(x2/p2) γ2=p2/ pϴ2 x2 По константе распределения (закон Нернста) Этот метод используется только в том случае, если вещество распределенное между двумя фазами α и β образует в одной фазе идеальный раствор, а в другой реальный. K=xДα/аД β аД β= xДα/К γД β= хД α/хДβ К По криоскопическим данным  𝑀1(Т,тв) 𝑀1(Т,тв)= 𝑀∅ + 𝑅𝑇𝑙𝑛а 1(𝑇,ж) 1 ∆𝐻𝑚,пл ∗ ∆Т −𝑙𝑛𝑎1 =  𝑅𝑇2пл Возьмем производную ∆𝐻𝑚, пл 𝑑∆𝑇 −𝑑𝑙𝑛𝑎1 = 𝑅𝑇2пл Вспомним 2 −оеуровнениеГиббса-Дюгема 𝑛1𝑑𝑀1 + 𝑛2𝑑𝑀2 = 0  1 𝑀1 = 𝑀∅ + 𝑅𝑇𝑙𝑛а1 2 𝑀2 = 𝑀∅ + 𝑅𝑇𝑙𝑛а2 𝑑𝑀1 = 𝑅𝑇𝑙𝑛а1 𝑑𝑀2 = 𝑅𝑇𝑙𝑛а2 𝑛1𝑑𝑙𝑛𝑎1 + 𝑛2𝑑𝑙𝑛𝑎2 = 0 −𝑛2 𝑑𝑙𝑛𝑎 = 𝑑𝑙𝑛𝑎 1 𝑛1 2 Подставляю (2)в(1) 𝑛2 𝑑𝑙𝑛𝑎 𝑛1 2 ∆𝐻𝑚, пл 𝑑∆𝑇 = 𝑅𝑇2пл𝑑𝑙𝑛𝑎2 = ∆𝐻𝑚,пл∗𝑛1 ∗ 𝑑∆𝑇 𝑅𝑇2пл∗ 𝑛2 𝑛2 = 𝑚2 (моляльность) {моль} кгр−ля 𝑛1 = 1000  М1 ∆𝐻𝑚,пл ∗ 1000  1 𝑑𝑙𝑛𝑎2 = 𝑅𝑇2пл 𝑀 ∗ 𝑚2 𝑑∆𝑇 𝑑𝑙𝑛𝑎2 = 𝑑∆𝑇 (3)  𝑘𝑓𝑚2 𝑘𝑓𝑚2Введем вспомогательную величину j=1-∆𝑇 𝑘𝑓∗𝑚2 -возьмемпроизводную 𝐾𝑓∆𝑇𝑑𝑚2 1 𝐾2𝑚2∗𝑚2 𝑑𝑗 = − 𝑘𝑓 ∗ 𝑚2𝑑∆𝑇 − 𝐾 + ∆𝑇𝑑𝑚2 −𝑑∆𝑇   𝐾2 ∗ 𝑚2 = 𝐾 𝑚 − 𝐾2 ∗ 𝑚2 = 𝐾 𝑚 −∆𝑇𝑑𝑙𝑛𝑚2  𝐾 𝑚 𝑓 2 𝑓 2 𝑓 2 dj=−𝑑𝑙𝑛𝑎2 + 𝑑𝑙𝑛𝑚2 − 𝑗 ∗ 𝑑𝑙𝑛𝑚2 𝑑𝑗 = −𝑑𝑙𝑛𝛾2 − 𝑑𝑙𝑛𝑛2 + 𝑑𝑙𝑛𝑚2 − 𝑗𝑑𝑚2 𝑚2 ∫𝑚2 𝑗 𝑚2 𝑗𝑑𝑚2 𝑚2=0 𝑑𝑙𝑛𝛾2 = − ∫0 𝑑𝑗 − ∫0 𝑚2 проинтегрируем 𝑙𝑛𝛾2 𝑚2  = −𝑗 − ∫ 𝑗 = −𝑗 − ∫ 𝑗𝑚2 0 𝑑𝑚 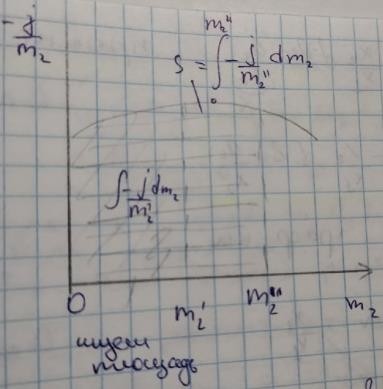 Определение активности растворителя (растворенного вещества) по активности растворенного вещества (растворителя) X1dϻ1+x2dϻ2=0 ϻ1=ϻ10+RT lnγ1+ RT lnx1 ϻ2=ϻ2ϴ+RT lnγ2+ RT lnx2 dϻ1=RTd lnγ1+ RT d lnx1 x1 lnγ1+x1 d lnx1 + x2 lnγ2+ x2 lnx2=0 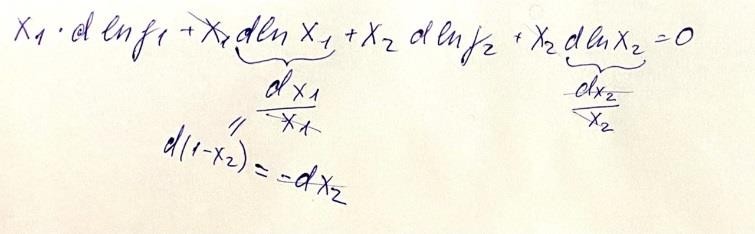 𝑑(1 − 𝑥2) = 𝑑𝑥2 X1 d ln γ1 + x2 d ln γ2 = 0  ∫𝑑 𝑙𝑛𝛾1 = ∫− 𝑥2 ∗ 𝑥1 ∗ 𝑑 𝑙𝑛 𝑎2 ∫𝑑 𝑙𝑛𝛾1 = ∫− 𝑥2 ∗ 𝑥1 ∗ 𝑑 𝑙𝑛 𝑎2𝑥2  ln γ1= ∫ − 𝑥2 𝑑 𝑙𝑛 𝑎2 ln γ1= ∫ − 𝑥2 𝑑 𝑙𝑛 𝑎2 𝑥1 𝑥2 𝑥1 𝑥2Правило фаз Гиббса (понятие фазы, вариантности системы, компонента системы).Правило фаз(или правило фаз Гиббса) —выражает основной закон фазового равновесия и устанавливает связь в равновесной системе между числом фаз (Ф), числом компонентов (К), которые присутствуют во всех фазах, и числом независимых термодинамических параметров (вариантностью системы С). Общее число параметров системы, характеризующих концентрацию, равно К • Ф. величина р, какого- либо компонента в данной фазе равна его химическому потенциалу во всех остальных фазах, то количество независимых уравнений для этого компонента равно Ф - 1. Очевидно, что для всех компонентов системы оно будет равно К(Ф - 1). Для каждой фазы существует одно уравнение состояния; значит, для системы число таких уравнений равно Ф. Таким образом, общее число уравнений, связывающих параметры, равно К(Ф - 1) + Ф. Разность между числом параметров и числом связывающих их уравнений равно числу независимых параметров системы (С). F=К – Ф +2 К- количество компонентов; Ф- количество фаз; F - Число степеней свободы; 2- T и P внешние факторы (С) число степеней свободы (F) возрастает с увеличением числа независимых компонентов (К) и уменьшается при росте числа фаз (Ф). При F = 0 в равновесии находится наибольшее число фаз. В общем случае, число фаз не может быть больше К + C (при этом F = 0): Ф К C. Например, для однокомпонентной (К = 1) системы при изменении двух внешних факторов температуры и давления (C = 2), максимальное количество фаз равно трем (твердая, жидкая и газообразная). Правило фаз Гиббса для однокомпонентной системы: в равновесной термодинамической системе, на которую из внешних факторов оказывают влияние только температура и давление, число термодинамических степеней свободы равно числу компонентов минус число фаз плюс два. F К Ф 2 Для двухкомпонентной системы (F = 2) диаграммы рассматривают без учета газовой фазы, то есть считают, что р = const, а изменяющимся внешним фактором считают только температуру(C=1). Тогда правило фаз Гиббса запишется следующим образом: С 2Ф 1 3Ф. Правилу фаз Гиббса не подчиняются коллоидные системы, в которых дисперсная фаза микроскопических размеров. Правило фаз не применимо к неравновесным метастабильным состояниям (переохлажденная вода, перегретый пар и т.д.). i При фазовом равновесии химические потенциалы каждого компонента равны во всех фазах (1)= i i (2)= ..= (p) i= 1, 2, .., C 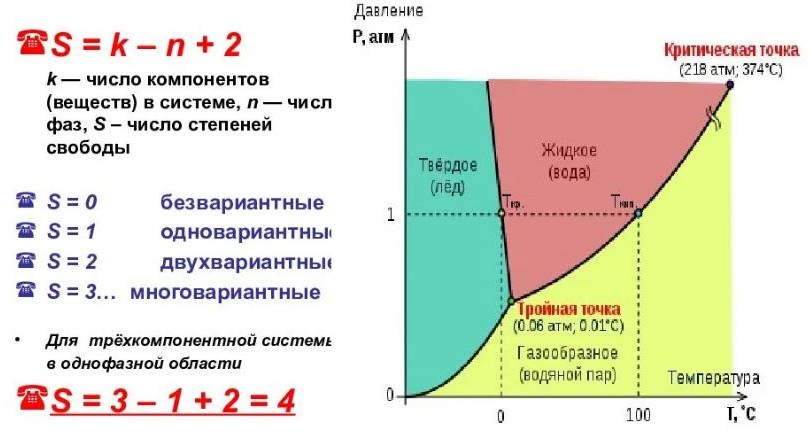  Термодинамическое условие стабильности фаз и фазового равновесия.В состоянии равновесия может находиться система, состоящая из нескольких различных по своим физико-химическим свойствам фаз, пространственно разделенных не изменяющимися с течением времени (или для квазиравновесного случая бесконечно медленно изменяющимися) границами раздела фаз. Если через эти границы не происходит макроскопический перенос, а сами фазы находятся в состоянии термодинамического равновесия, то такая термодинамическая система, несмотря на свою неоднородность, будет находиться в состоянии термодинамического равновесия. условие не обязательно соответствует равенству давлений и с разных сторон границы раздела, так как сама эта граница, в случае, если её форма не представляет собой плоскости, может создавать дополнительное межфазное давление имеет вид:  где: - дополнительное давление на первую фазу, создаваемое границей её раздела со второй. Если считать границы раздела фаз плоскими, то условие станет эквивалентным предположению о равенстве давлений по обе стороны где: - дополнительное давление на первую фазу, создаваемое границей её раздела со второй. Если считать границы раздела фаз плоскими, то условие станет эквивалентным предположению о равенстве давлений по обе стороны. Термодинамическим критерием фазового превращения – является неравенство химических потенциалов; вещество из фазы с большим хим потенциалом будет самопроизвольно переходить в фазу с меньшим хим потенциалом  Диаграммы состояния в однокомпонентной системе. Уравнение Клаузиуса-Клайперона.В состоянии фазового равновесия: ϻα =ϻβ при Т, Р=CONST (1) При изменении температуры на б.м. величину d T, давление на б.м. dp, устанавивается новое фазовое равновесие ϻα +d ϻα =ϻβ + d ϻβ (2) Из (1) и (2) следует d ϻα= d ϻβ Т.k. ϻ=g то : ϻα =-Sαd T+ Vαdp ϻβ=-Sβd T+ Vβdp → -Sαd T+ Vαdp=-Sβd T+ Vβdp   → 𝒅𝒑 = ∆𝑺ф.п. = ∆Н𝒎,ф.п. → 𝒅𝒑 = ∆𝑺ф.п. = ∆Н𝒎,ф.п.𝒅𝑻 ∆𝑽ф.п. Т∆𝑽ф.п. 𝒅𝒑=∆Н𝒎,ф.п.Уравнение Клайперона 𝒅𝑻 Т∆𝑽 𝒅𝑻 Т∆𝑽тв→ж  𝒅𝒑= ∆Н𝒎,пл 𝒅𝒑= ∆Н𝒎,пл𝒅𝑻 Т(𝑽ж−𝑽тв) Каждая точка лежащая на (о-а) характеризует равновесие 2х фаз тв и ж К1=1-2+2=1 система моновариантна К2=1-1+2=2 система бивариантна ж→п  𝒅𝒑= ∆Н𝒎,исп 𝑽п ≫ 𝑽ж 𝒅𝒑= ∆Н𝒎,исп 𝑽п ≫ 𝑽ж𝒅𝑻 Т(𝑽п−𝑽ж) Каждая точка лежащая на (о-в) –равновесие двух фаз ж и п Для ф.п. ж-п для высоких V и T существует особая точка, которая называется критической В критическом положении ж и п фазы тождественны и они переходят друг в друга без границы раздела, без мениска В области крит. Т. Вещество обладает уникальными свойствами. Оно имеет сжимаемость как у газа, а плотность как у жидкости В кр. Т. Fк= 1-2+2-1=0 (на нее повлиять нельзя) тв→п (возгонка, сублимация) 𝒅𝒑= ∆Н𝒎,субл > 𝟎𝒅𝑻 Т(𝑽п−𝑽тв) Т. О – называется тройной. В ней в равновесии 3 фазы одновременно К0=1-3+2=0 (ей также нельзя управлять) Наклон кривых ОА, ОВ, ОС на плоской диаграмме состояния определяется знаком и величиной производной dP/dT, выражаемой уравнением Клапейрона-Клаузиуса. Определение термодинам. Хар-к фаз. Перехода ж-п 𝒅𝒑=∆Н𝒎,исп𝒅𝑻 Т(𝑽п) При переходе из уравнения Клаперона в ур Калузиуса-Клаперона: Vmп>>Vmж → Vmж пренебрегаем, след. Vп=Vж Считаем, что пар ведет себя как идеальный газ и можно использовать ур-е состяния идеального газа: Pv=RT→V=RT/p Считаем, что мольная энтальпия испарения (Нm.исп) является независимой от Т След. 𝒅𝒑=∆Н𝒎,исп∗𝒑 →𝒅𝒑=∆Н𝒎,исп∗𝒑 𝒅𝑻𝒅𝑻 𝑹𝑻𝟐 𝒑 𝑹𝑻𝟐  Последовательная Реакция первого порядка с общим реагентом Две стадии называются последовательными, если вещество, образующееся в первой стадии, является исходным реагентов во второй. Если обе стадии первого порядка, то мы имеем дело с последовательной реакцией первого порядка:  При этом по условию материального баланса сумма текущих концентраций всех участников реакции равна начальной концентрации исходного реагента: [A] + [B] = [C] = [A]0 (4) Наша задача – выразить в явном виде концентрации всех веществ: реагента А, промежуточного продукта В, конечного продукта С. Что касается реагента А, то затруднений нет, поскольку дифференциальное уравнение (1) совпадает с уравнением простой реакции первого порядка. Это уравнение мы не раз интегрировали:     Как видно из представленных графических зависимостей, концентрация реагента А изменяется по классической экспоненте, концентрация продукта С монотонно возрастает, стремясь к начальной концентрации реагента. Зависимость концентрации промежуточного продукта В имеет вид кривой с экстремальной точкой М, которой соответствует максимальная концентрация вещества [В]. Скорость накопления вещества В в точке М равна скорости его расходования, а общая скорость изменения концентрации В равна нулю. Найдем координаты точки М, для чего продифференцируем уравнение (13) и приравняем дифференциал к нулю. 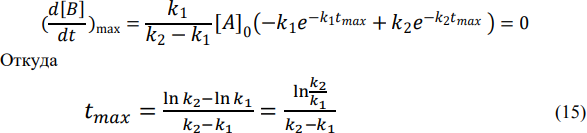  Приближенные методы в анализе кинетических схем хим реакций. Метод Боденштейна и квазиравновесного приблежения. Для большинства реакций, содержащих несколько элементарных стадий, кинетические уравнения могут быть достаточно сложными. Вместе с тем, константы скорости отдельных стадий могут сильно различаться, что позволяет использовать при решении кинетических уравнений приближенные методы. Наиболее часто используется метод квазистационарных концентраций (метод Боденштейна) Анализ уравнений (15) и (16) показывает, что при k2>>k1 и при увеличении соотношения k2/k1 значение [В]мах и время его достижения (tмах) уменьшаются. Это можно показать непосредственно расчетным методом. Зададим в последовательной реакции первого порядка [A]0 = 0,1 M, k2 = 1,0 c-1 , k1 = 0,1 c-1 , т.е. различие в константах скорости не слишком большое k2/k1 = 10. Подставляем эти значения в уравнения (15) и (16). А теперь предположим, что константы скорости второй и первой стадий отличаются в 1000 раз, т.е. промежуточное вещество В образовалось в первой стадии и, являясь очень реакционноспособным, сразу же превращается в продукт С: [A] 0 = 0,1 M, k2 = 100 c-1 , k1 = 0,1 c-1 , т.е. различие в константах скорости большое k2/k1 = 1000. Как видно из расчетов, чем более реакционноспособным является промежуточное вещество, тем меньше его максимальная концентрация и тем меньше время достижения этой концентрации:  Подобный анализ позволяет в первом приближении считать, что скорость накопления промежуточного продукта при условии его высокой реакционной способности равна 0. Другими словами, промежуточное вещество образуется в первой стадии с некоторой константой скорости k1 и сразу же в силу своей высокой реакционной способности вступает во вторую стадию с существенно большей скоростью. В этом физическая основа метода квазистационарного приближения, или Боденштейна. В связи с чем концентрация промежуточного вещества в любой момент времени мала (но не равна нулю), а, следовательно, мала и скорость ее изменения, которую принимают равной нулю. Так, в соответствии с методом квазистационарного приближения скорость накопления промежуточного продукта B при условии его высокой реакционной способности равна приближенно нулю: d[В]/dt≈ 0 . Поскольку с математической точки зрения нулю равна производная от постоянной величины, то это приближение часто называют стационарным. В качестве примера использования этого метода рассмотрим следующую схему реакции: В соответствии со схемой (17) скорость реакции определяем по накоплению продукта П. Продукт образуется в элементарной стадии с константой скорости k2. Поскольку на основании постулатов химической кинетики каждая элементарная стадия сложной реакции протекает независимо и к каждой элементарной стадии можно применить основной постулат химической кинетики, то В конечном виде кинетического уравнения не должно быть никаких промежуточных продуктов, т.е. должны быть только концентрации реагентов. В связи с чем, все приближенные методы анализа кинетических схем направлены на то, чтобы так или иначе выразить концентрации промежуточных продуктов через концентрацию исходных веществ. В методе Боденштейна задача сводится к тому, чтобы правильно написать скорость накопления промежуточного продукта. Каждую элементарную стадию схемы считаем независимой и к каждой стадии применяем основной постулат химической кинетики. Так, в соответствии со схемой реакции вещество В образуется на стадии с константой скорости k1. Скорость образования на основании основного постулата химической кинетики равна k1[А]. Расходуется промежуточное вещество В на стадии с константой скорости k-1, скорость убыли вещества В на этой стадии равна - k-1[В]. Также вещество В расходуется на элементарной стадии с константой скорости k2. Скорость убыли вещества В на этой стадии равна –k2[В]. Тогда суммарная скорость накопления промежуточного продукта В будет слагаться из скорости его образования в первой стадии и скоростей его убыли во второй стадии и стадии, обратной первой. И эта скорость в методе квазистационарного приближения равна приблизительно нулю: Поскольку правая часть уравнения (18) стала определенной, равной 0, то уравнение (18) можно решить относительно неизвестной концентрации промежуточного продукта: Другим приближенным методом в химической кинетике является метод квазиравновесного приближения. В квазиравновесном приближении принимается, что равновесие в обратимых стадиях достигается значительно быстрее, чем промежуточные соединения вступают в дальнейшие превращения (k-1 >> k2). равна практически равновесной, определяемой уравнением , (20) где [В]квазир, [А]квазир – квазиравновесные концентрации в данный момент времени; K– константа равновесия первой обратимой стадии. Квазиравновесные концентрации – это такие концентрации реагентов, которые определяются константой равновесия, но не в равновесном, а в любом другом моменте времени. В методе квазиравновесного приближения концентрацию промежуточного продукта В выражаем через константу химического равновесия в соответствии с уравнением (20): [В]квазир = К [А]квазир, где [А]квазир равна аналитически определяемой концентрации реагента А. Выражая концентрацию промежуточного соединения В через константу равновесия, имеем V=k2 K[A]=(k2k1/k-1)*[A]. (21) Задача. Используя метод Боденштейна, найдите уравнение скорости образования фосгена (COCl2) согласно следующему механизму: |